New Era of Management Concept on Pulmonary Fibrosis with Revisiting Framework of Interstitial Lung Diseases
Article information

Abstract
The disease concept of interstitial lung disease with idiopathic pulmonary fibrosis at its core has been relied on for many years depending on morphological classification. The separation of non-specific interstitial pneumonia with a relatively good prognosis from usual interstitial pneumonia is also based on the perception that morphology enables predict the prognosis. Beginning with dust-exposed lungs, initially, interstitial pneumonia is classified by anatomical pathology. Diagnostic imaging has dramatically improved the diagnostic technology for surviving patients through the introduction of high-resolution computed tomography scan. And now, with the introduction of therapeutics, the direction of diagnosis is turning. It can be broadly classified into to make known the importance of early diagnosis, and to understand the importance of predicting the speed of progression/deterioration of pathological conditions. For this reason, the insight of “early lesions” has been discussed. There are reports that the presence or absence of interstitial lung abnormalities affects the prognosis. Searching for a biomarker is another prognostic indicator search. However, as is the case with many chronic diseases, pathological conditions that progress linearly are extremely rare. Rather, it progresses while changing in response to environmental factors. In interstitial lung disease, deterioration of respiratory functions most closely reflect prognosis. Treatment is determined by combining dynamic indicators as faithful indicators of restrictive impairments. Reconsidering the history being classified under the disease concept, the need to reorganize treatment targets based on common pathological phenotype is under discussed. What is the disease concept? That aspect changes with the discussion of improving prognosis.
Dawn of Interstitial Lung Disease Diagnosis
Beginning of the diagnosis of interstitial lung disease was begun with pneumoconiosis of quarryers in Europe [1]. Virchow has been a professor of pathology at the Humboldt-Universität zu berlin since 1956 and laid the foundation for human pathology. Furthermore, Liebow [2] has classified interstitial pneumonia and pulmonary fibrosis based on the lung anatomy. At that time, many interstitial pneumonia/pulmonary fibrosis was the collective term for pneumoconiosis due to coal lung mining with superior upper lobe [3].
Later, Katzenstein and Fiorelli [4], who found a form different from that of usual interstitial pneumonia (UIP), with a uniform temporal phase and a favorable prognosis, proposed to distinguish it as nonspecific interstitial pneumonia/fibrosis.
At that time, treatment and management recommendations were not addressed in these tones, and were solely aimed at classification and discrimination. In addition, pathological forms with good responsiveness to corticosteroids (CS) treatment were taken up as bronchiolitis obliterans organizing pneumonia [5].
The Dawn of Therapeutics
In 1999, the therapeutic effect of interferon gamma (IFNg) on idiopathic pulmonary fibrosis (IPF) was reported [6]. This is the first paper on the possibility of treatment and management of IPF. Nine patients with IPF treated with interferon gamma 1b or as same number for placebo reveled significant improvement of total lung capacity and oxygen concentration in blood at rest. However, it has been found that IPF is a heterogeneous population in response to treatment with IFNg (Figure 1) [7,8].
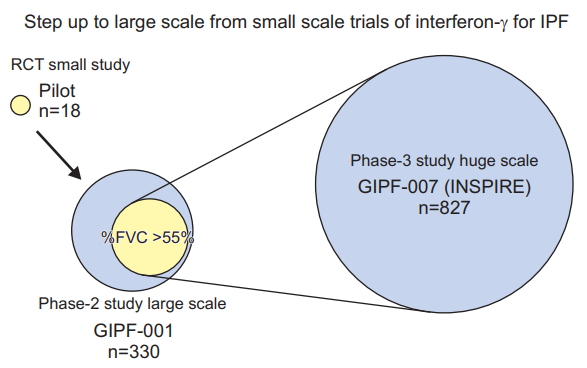
Diversity of background characteristics affecting failed result of primary endpoint in interferon gamma trials for idiopathic pulmonary fibrosis. IPF: idiopathic pulmonary fibrosis; RCT: randomized clinical trial; FVC: forced vital capacity. Adapted from Azuma and Usuki. Respir Med CME 2008;1:75-81, with permission of Elsevier [12].
Numerous treatment trials have been attempted, most of which have failed. At that time, the optimal metrics for treatment were not authorized. Therefore, the evaluation indices of clinical trials were various and considered to be one of the reasons for failure of clinical trials.
1. Pirfenidone clinical trials
In the meantime, we looked for a time-lapse follow-up test in the United States [9] and sought a way to evaluate drug efficacy in a shorter time. As a result, we considered that the evaluation by comparing the oxygen consumption, which incorporates the ability to support constant velocity exercise using a treadmill, could be a better index. Therefore, we conducted a search for the effectiveness of pirfenidone for IPF [10]. As a result, while there were cases in which the improvement of oxygen availability could be evaluated over time (Figure 2), there were cases in which the improvement was impossible, and it was difficult to establish a method for evaluating clinical trials. Rather, we set a highly reproducible vital capacity (VC) as the primary endpoint and planned a new pirfenidone verification test to prove the effectiveness of IPF in reducing VC [11].

Two examples who responded to pirfenidone and improved oxygen desaturation area during the study. Oxygen desaturation lines measured every 3 months, came up to the value at rest along time periods. These are thought to be good responder. SpO2: saturation of pulse oximetry.
Large scale studies were also conducted in Europe and the United States with forced vital capacity (FVC) set as the primary endpoint. One of the twin trials met the primary endpoint, but the other did not meet.
One of the reasons is that IPF patients enrolled in clinical trials include many cases with emphysema complications, and the placebo group did not show a significant decrease in FVC, which resulted in an unprovable pirfenidone efficacy [13]. In the next planned ASCEND (A Study of Cardiovascular Events in Diabetes) trial, IPF patients meeting the restriction of forced expiratory volume in 1 second >70% were registered, and the comparison of the FVC decline rate of IPF patients without emphysema was examined, and the primary endpoint meet was successfully achieved [14]. IPF is a very heterogeneous disease unit in time and space.
2. Corticosteroid benefits (PANTHER trial)
IPF has long used CS as a symptomatic treatment. However, as a result of the PANTHER test designed to evaluate the efficacy of CS, N-acetylcysteine, and azathioprine, it was concluded that ‘CS is harmfull.’ Looking at the details of the results, the reason was that the frequency of hospitalization and death increased significantly in the CS combination group compared to the placebo control group [15]. Analysis of the entry protocol shows that the course of CS decreasing up to 15 weeks, at which hospitalization and death increases, is extremely fast compared to CS in the IFNg trial. In my understanding, the schedule for the CS decreasing process has a very significant impact (Figure 3).

Therefore, the above did not conclude the pros and cons of low-dose CS. The usefulness of CS for populations that are not easily distinguished in daily clinical practice, such as unclassifiable idiopathic interstitial pneumonias (IIPs) and interstitial pneumonia with autoimmune features (IPAF), suggesting the involvement of immunological conditions. Furthermore, the combination of anti-fibrotic drugs with CS should be evaluated.
3. Nintedanib trials for IPF (TMORROW, INPULSIS-1, -2, and INPULSIS-ON)
Nintedanib is the next anti-fibrotic drug treatment trial for IPF that has been enthusiastically started around 2010. The TMORROW study was conducted as a dose-finding study, and the recommended dose was 150 mg twice daily [16]. The INPULSIS-1 and -2 tests, which were conducted as proof of concept tests, were conducted in 13, 17 countries and 98, 107 facilities, respectively, and ended in October 2013 (Table 1). As a result, the annual reduction in FVC was reduced to about 50% in both tests. Based on the results of this clinical trial, Pharmaceuticals and Medical Devices Agency (PMDA) approved production and clinical use following U.S. Food and Drug Administration (FDA) and European Medicines Agency approvals [17]. In the INPULSIS-ON study, which was continued as a post-marketing extension study, some cases were having treatment duration for reached 5 years, and as a mid-term report, suppression of FVC reduction was maintained in active treatment changed from the placebo group and in the continuation group from active treatment and of these treatment group expressed the time to first acute exacerbation was significantly prolonged [19].
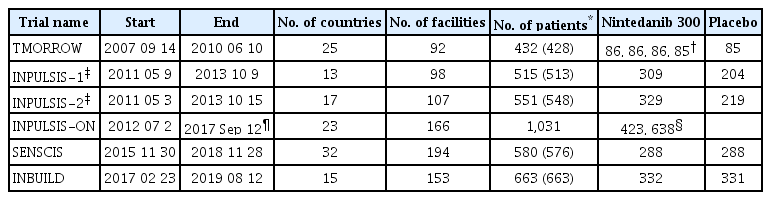
Comparison of randomized control trials of nintedanib
Based on the summary assessment of the INPULSIS trials, a reduction in FVC of 24 weeks could not predict a decrease in FVC of the next 24 to 52 weeks, but could predict mortality. Based on these results, we need to understand that blocking an FVC% >10% decrease is directly linked to improving the prognosis of IPF patients [20].
Non-IPF Treatment Trials: SENSCIS, INBUILD, INMARK (Biomarker), PFD for Unclassifiable Interstitial Lung Disease
Because of the poorest prognosis of interstitial lung disease (ILD), IPF has become a target for new drug development, and many therapeutic drug developments have been attempted. But is non-IPF good? The answer is no. The IPF international diagnostic guidelines exclude similar ILDs such as IPAF and Unclassifiable IIPs in order to accurately diagnose IPF, but the prognostic curve is never good [21]. In other words, even if the disease concept is different, the pathology presenting a UIP pattern has a poor prognosis. In recent years, these treatment trials have been planned and randomized clinical trials have been conducted. The SENSCIS trial is one of them, in which clinical trials have been developed with an unprecedented number of cases for systemic sclerosis (SSc-ILD). As a result, SSc-ILD within 7 years of diagnosis was recruited to clinical trials, and the annual FVC decline in the nintedanib group was halved compared to the placebo group [22]. FVC was suppressed by the combined use of mycophenolate, but the suppression of nintedanib was an additive effect, and the combined use also ensured the safety. However, no significant difference was found between the two groups for modified Rodnan skin score and Saint George’s Respiratory Questionnaire as secondary endpoints. We proved that nintedanib suppressed not only IPF but also Fc reduction of SSc-ILD, and now FDA and PMDA have approved, and clinical treatment prescription was enabled on December 20, 2019.
In addition, the inhibitory effect of nintedanib was examined for re-categorized disease groups using a common disease behavior called progressive fibrosis. The following criteria were applied to the progressive entry criteria.
Patients were required to meet one of the following criteria for ILD progression in the 24 months before screening, despite management:
(1) Relative decline in FVC ≥10% predicted, (2) Relative decline in FVC ≥5 to <10% predicted and worsened respiratory symptoms, (3) Relative decline in FVC ≥5 to <10% predicted and increased extent of fibrosis on high-resolution computed tomography (HRCT), and (4) Worsened respiratory symptoms and increased extent of fibrosis on HRCT. Fifty percent (1), 30% (2 and 3), and 20% (4) were included in the study, respectively.
As a result, treatment with nintedanib significantly suppressed the decrease in FVC, and this effect was the result of obtaining a bimorph with IPF and SSc-ILD [23].
On the other hand, in pirfenidone treatment studies, studies have been conducted on unclassifiable IIPs to study the suppression of progression of progressive fibrosis. Similarly, the result of suppressing the decrease in FVC has been obtained [24]. For the time being, it has been suggested that suppression of a decrease in FVC may lead to improved prognosis.
Is IPF a Disease Concept? Thinking Based on Disease Behavior: Reconsideration of Disease Concept, Thinking Change of Functional Impairment from Definition by the Shape
It is time to rethink “What is a disease concept?” In the past, there is a history of morphological classification. Katzenstein, who separated non-specific interstitial pneumonitis from IPF, noted its good prognosis and differences in treatment, and suggested excluding it from conventional IIPs [5]. However, the need for early diagnosis makes it more difficult to distinguish IPF from other IPs. The prognosis of ILA on diagnostic imaging is worse than the group without ILA [25]. Even with varying degrees of poor prognosis, does a lung with ILA need to continue to identify opportunities for intervention?
IPF/UIP is the disease concept with the worst prognosis from a pathological and diagnostic point of view. However, comparing the annual decline rate of FVC, which is a prognostic surrogate marker, the decrease in FVC of IPF is various and not uniform. Many researchers acknowledge that the behavior of the disease itself is not linear, but rather diverse [26].
Based on treatment discussions, the search for biomarkers for prognostic factors has begun. What is the marker that predicts the decline in FVC? CRPM is a strong candidate [27]. However, it was not promising for nintedanib evaluation of treatment [28].
In addition to predicting FVC decline, predicting diffusing capacity for carbon monoxide (DLco) decline may be even more important. Predicting advanced pulmonary arterial hypertension mergers is important as a prognostic indicator. Increased gene expression of Periostin correlates with a decrease in DLco, implying a potential prognostic value [29]. Furthermore, complications such as “acute exacerbation” and “complications with lung cancer” greatly influence the prognosis. Risk reduction of complications can significantly affect prognosis. We believe that ‘Risk reduction of complications’ can be a therapeutic target in the future.
The prognosis of IPF, a chronic respiratory disease, depends on various factors. In the era of steroid use, even if the symptoms were alleviated, infections were likely to be induced, which might have worsened the prognosis of life.
In recent years, scientific evidence has gradually accumulated, but regarding treatment choices for IPF patients, not only evidence based on scientific medicine, but rather ‘shared decision making (SDM)’ using patient’s own values based on disclosure of evidence-based medicine (EBM) information leading to SDM is recommended. It is necessary to select treatments that respect individual patient values with empathy. EBM needs SDM, and SDM needs EBM. Patients need both [30,31].
Notes
Authors’ Contributions
Conceptualization: Azuma A. Methodology: Azuma A. Formal analysis: Azuma A, Richeldi L. Data curation: Azuma A, Richeldi L. Validation: Azuma A. Investigation: Azuma A. Writing - original draft preparation: Azuma A. Writing - review and editing: Richeldi L. Approval of final manuscript: all authors.
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Funding
No funding to declare.