Subphenotypes of Acute Respiratory Distress Syndrome: Advancing Towards Precision Medicine
Article information

Abstract
Acute respiratory distress syndrome (ARDS) is a common cause of severe hypoxemia defined by the acute onset of bilateral non-cardiogenic pulmonary edema. The diagnosis is made by defined consensus criteria. Supportive care, including prevention of further injury to the lungs, is the only treatment that conclusively improves outcomes. The inability to find more advanced therapies is due, in part, to the highly sensitive but relatively non-specific current syndromic consensus criteria, combining a heterogenous population of patients under the umbrella of ARDS. With few effective therapies, the morality rate remains 30% to 40%. Many subphenotypes of ARDS have been proposed to cluster patients with shared combinations of observable or measurable traits. Subphenotyping patients is a strategy to overcome heterogeneity to advance clinical research and eventually identify treatable traits. Subphenotypes of ARDS have been proposed based on radiographic patterns, protein biomarkers, transcriptomics, and/or machine-based clustering of clinical and biological variables. Some of these strategies have been reproducible across patient cohorts, but at present all have practical limitations to their implementation. Furthermore, there is no agreement on which strategy is the most appropriate. This review will discuss the current strategies for subphenotyping patients with ARDS, including the strengths and limitations, and the future directions of ARDS subphenotyping.
Introduction
Acute respiratory distress syndrome (ARDS) is defined as the acute onset of non-cardiogenic bilateral pulmonary edema causing hypoxemia. The Berlin definition, established in 2012, is used to both define and grade the severity of ARDS [1]. This standardized definition amalgamates patients with a variety of different etiologies, clinical manifestations, responsiveness to therapies, levels of acute illness, and radiographic patterns. On one hand, this approach has optimized and standardized enrollment in large muti-center ARDS clinical trials and contributed to important advances in the care we provide to patients with ARDS, including low tidal volume ventilation, positive end expiratory pressure (PEEP), and extracorporeal membrane oxygenation. Yet at the same time, innumerable clinical trials of drugs that appeared highly promising in pre-clinical and early phase clinical studies have failed to identify effective pharmacotherapies for ARDS, likely due at least in part to the underlying heterogeneity of the syndrome. Dividing ARDS into subphenotypes, or distinct subgroups characterized by specific and consistent observable traits, may allow investigators to apply precision medicine to the field of ARDS and target the right therapy for the right patient. The identification of more homogenous subgroups of patients with ARDS based on observable traits lends itself to both predictive and prognostic enrichment, more efficient randomized control trials, and the detection of a treatment effect if one were to exist. An analogous “precision medicine” approach has revolutionized and advanced the search for novel and effective pharmacotherapies in cystic fibrosis, cancer, and asthma, among others. This review outlines strategies currently being used to identify subphenotypes of ARDS and discusses future directions in ARDS research, including the potential for future incorporation of subphenotypes into clinical trials and bedside care of ARDS.
ARDS Background
ARDS was first described in 1967 by Ashbaugh et al. [2] who observed 12 patients with acute onset of “tachypnoea, hypoxaemia, and loss of compliance after a variety of stimuli.” They noted that the inciting event for each of these patients varied, but that their clinical presentation was similar in the development of alveolar edema, hypoxemia, poor responsiveness to traditional respiratory therapy, and clinical improvement with PEEP [2]. In the decades that followed, the pathogenesis and the pathobiology of ARDS have been better understood. Recent reviews have summarized our current understanding of ARDS pathogenesis in detail [3,4]. Briefly, direct or indirect injury to the lungs initiates an inflammatory cascade mediated largely by neutrophils and macrophages, leading to injury of the alveolar-epithelial barrier and the subsequent filling of the alveolar spaces with protein-rich edema fluid, which leads to poorly ventilated areas of lung, increased physiologic dead space, and severe hypoxemia [5].
In 1994, the American-European Consensus Conference attempted to impart structure and clarity into the confusion behind the definition of ARDS [6]. By 2012 the Berlin Criteria had replaced the AECC criteria and included (1) onset within 7 days of an insult, (2) hypoxemia with a PaO2/FiO2 (P/F) ratio of ≤300, (3) non-cardiogenic pulmonary edema, and (4) bilateral infiltrates [1]. The intent of these definitions was to codify a condition that left epidemiologists, clinicians, and researchers vexed by the perception of significant heterogeneity. However, these highly sensitive diagnostic criteria have continued to capture a clinically and biologically heterogeneous set of patients. In contrast, in asthma and cancer, the concept of biological and clinical phenotypes and subphenotypes has been utilized to rapidly advance patient care. In breast cancer, for example, the development of trastuzamab to target patients whose tumors were positive for the transmembrane receptor human epidermal growth factor receptor 2 revolutionized the standard of breast cancer care [7]. The recognition that asthma is a heterogenous disease composed of numerous clinical and biological phenotypes and subphenotypes paved way for a more comprehensive understanding of the pathobiology and the development and use of immunomodulator therapy in asthma [8,9]. While most landmark clinical trials that guide the current management of patients with ARDS have treated all patients similarly, an anthology of work has focused on subphenotypes of ARDS.
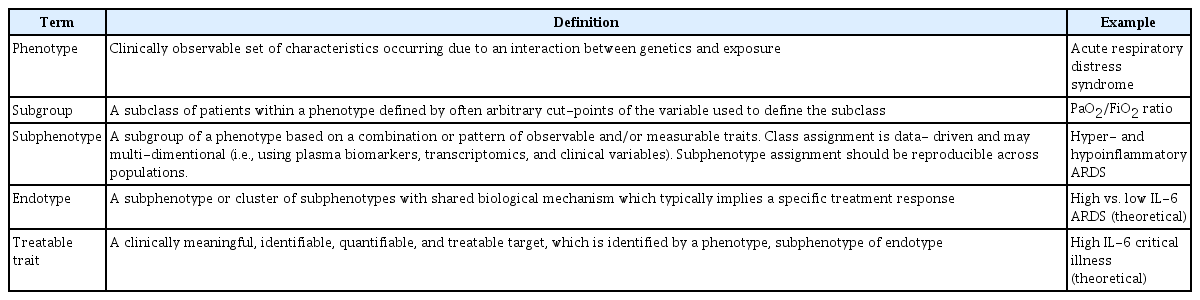
Defining the Terms Phenotypes, Subphenotypes, Endotypes, Treatable
Terminology in this field remains under some debate; however, a recent European Society of Intensive Care Medicine (ESICM) guideline on ARDS provides one approach to terminology which we will utilize in this review (Table 1) [10]. It is important to note, that based on the ESICM guidelines, that ARDS itself is a phenotype, a clustering of patients based on clinically observable traits that occur as a result of the interaction between host and environment. A combination or pattern of traits defines a subphenotype. The various approaches used to subphenotype patients with ARDS will be the subject of this review (Figure 1).

A clinically observable set of traits is the definition of a phenotype. Acute respiratory distress syndrome (ARDS) represents a distinct phenotype. Subphenotypes of ARDS are based on a combination or pattern of observable traits. The figure above depicts both unidimensional and multidimensional strategies to define subphenotypes of ARDS. This figure is created with BioRender.com. P/F: PaO2/FiO2; COVID-19: coronavirus disease 2019; PEEP: positive end expiratory pressure; sRAGE: soluble receptor for advanced glycation end product; vWF: Von Willebrand factor; TNF-r1: tumor necrosis factor-receptor 1; IL: interleukin; IFN-γ: interferon γ; ANG1/2: angiopoietin 1/2; PAI-1: plasminogen activator inhibitor 1.
Clinical Based Subphenotypes
1. Etiology
The diverse set of etiologies that can cause ARDS contributes significantly to the heterogeneity of the syndrome. For example, patients who experience ARDS after major trauma have a much better survival (20% mortality) than their non-trauma counterparts (>40% mortality) [11]. Furthermore, in trauma, the development of ARDS does not seem to worsen the mortality beyond the initial traumatic insult [12]. Trauma ARDS patients may represent a meaningfully different subgroup of ARDS with a distinct pathophysiology and clinical course.
In recent years, numerous attempts have been made to differentiate coronavirus disease 2019 (COVID-19) and non-COVID-19 ARDS. This distinction remains controversial. This phenotyping schema originated from the observation that some COVID-19 ARDS patients had physiology that differed from “classical ARDS” including severe hypoxemia with “near” normal lung compliance, low lung weight, and low lung recruitability. This subphenotype was labeled “Type L” in contrast to the more classical ARDS known to have high elastance, high lung weight, and high recruitability, termed “Type H” [13]. However, in a retrospective analysis of COVID-19 patients, it was often impossible to distinctly divide patients into the “Type L” or the “Type H” subphenotype, with most patients experiencing features of both [14]. In a single-center comparison of inflammatory biomarkers and clinical outcomes from COVID-19 patients to classical non-COVID-19 ARDS, Bain et al. [15] demonstrated that both groups of patients had similar inflammatory cytokines and 60-day mortality. Bos et al. [16] sought to determine if there were distinct subphenotypes of COVID-19 ARDS. Latent class analysis (LCA) composed of respiratory and ventilator data after the initiation of mechanical ventilation identified only a single class, effectively refuting the hypothesis that distinct “L” and “H” subphenotypes of COVID-19 ARDS exist [16].
ARDS can alternatively be subdivided into direct or indirect ARDS: specifically, direct ARDS occurs when the acute insult directly impacts the lungs (e.g., pneumonia, aspiration, inhalation), and indirect ARDS results from non-pulmonary sources (e.g., non-pulmonary sepsis and pancreatitis). Akin to results from animal models, indirect ARDS in humans is characterized by more endothelial damage, while epithelial damage is a hallmark of direct ARDS [17]. In contrast, Morisawa et al. [18] noted that patients with indirect ARDS had lower pulmonary vascular permeability index and extravascular lung water index when compared to direct ARDS. Some authors have identified differences in pulmonary physiology and radiographic findings between these two subphenotypes. In one study, lung elastance was higher in patients with direct ARDS, whereas elastance of the chest wall was more than two-fold higher in the indirect ARDS patients19. Increasing PEEP in direct ARDS patients worsened lung compliance but resulted in alveolar recruitment in indirect ARDS patients [19]. Despite these differences in cytokines and physiology, a large meta-analysis indicated that there was no difference in mortality when comparing direct to indirect ARDS [20]. Furthermore, bedside classification is often challenging, as many patients will have numerous potential ARDS risk factors. Clinical trials have also failed to demonstrate consistent differences in response to treatment on the basis of direct versus indirect etiology of lung injury. Thus, the clinical impact of this distinction is limited at present.
2. Timing
Several groups have attempted to divide ARDS based on the timing of onset. In 1999, Croce et al. [21] defined “early-onset” and “late-onset” ARDS within the post-trauma ARDS population. Early ARDS, defined as disease within 48 hours of hospital admission, was more frequently associated with hemorrhagic shock. Late ARDS was associated with blunt injury, pneumonia, systemic inflammatory response, and multiorgan system failure [21].
When LCA was applied to a cohort of patients with post-trauma ARDS, two classes were identified, the “early-onset” and “late-onset,” similarly divided at the 48-hour cutoff. The early-onset class required significantly more red blood cell products and had a lower systolic blood pressure. However, there was no difference in mortality between the two groups. Plasma biomarkers obtained at the time of enrollment revealed a significantly higher level of angiopoietin 2 (Ang-2) and the soluble receptor for advanced glycation end product (sRAGE) in the early-onset class consistent with more significant impairment of the alveolar capillary barrier [22]. Applying the same early versus late-onset construct to allcomers with ARDS (i.e., not restricted to trauma induced ARDS), Zhang et al. [23] found that late-onset ARDS patients had a higher mortality and were more likely to die sooner than early-onset.
3. Physiology
Physiological parameters beyond the P/F ratio have been considered to assign patients with ARDS into subgroups. These parameters have been linked to outcomes in ARDS and, like the P/F ratio, may allow for enrichment of clinical trials. Driving pressure, or the difference between the plateau pressure and the PEEP, represents the variation in positive pressure that the lung parenchyma is exposed to during each ventilatory cycle. Driving pressure is strongly associated with mortality in ARDS, with the risk of death increasing as it exceeds 15 cmH2O [24]. Dead space fraction, and ventilatory ratio as a surrogate for dead space fraction, are also both associated with mortality in ARDS [25-27]. For every 0.05 increase in the dead space fraction, there was a 45% increase in the odds of death [25]. When patients with ARDS were stratified based on a ventilatory ratio of greater than or less than two, mortality was significantly higher with a ventilatory ratio greater than or equal to two [26]. Combining dead space and P/F ratio may allow categorization of patients with ARDS into those with ventilation impairment, oxygenation impairment, or both. These physiological parameters have yet to be used to enrich clinical trials (though the PRACTICAL platform led by Canadian investigators is planning such an approach; https://practicalplatform.org/) but offer a novel subgrouping strategy that may be particularly useful in trials exploring strategies of mechanical ventilation.
4. Radiographic patterns
The radiographic presence of bilateral opacities not otherwise explained by pleural effusions, lobar/lung collapse, or nodules is a component of the Berlin definition [1]. Puybasset et al. [28] described three groups of patients with distinct radiographic findings with ARDS, subsequently labeled “diffuse attenuation,” “lobar attenuation,” and “patchy attenuation.” These patients had different volumes of lung gas and healthy tissue [28]. These groups were subsequently consolidated into “focal” and “non-focal” ARDS and have been reported to have a differential response to recruitment maneuvers [29]. Patients with focal attenuation appear to be less responsive to recruitment maneuvers and open lung ventilation, with less improvement in PaO2 and lung aeration, and have a lower mortality compared to non-focal ARDS [29]. Plasma sRAGE, a marker of epithelial injury and alveolar fluid clearance, is also higher in patients with non-focal ARDS [30], offering a possible biological mechanism to explain the observed radiographic heterogeneity.
Kim et al. [31] explored responsiveness to prone positioning in focal versus non-focal ARDS and found no difference in the change in P/F ratio with proning between the two radiographic subphenotypes. Radiographic patterns are an enticing subphenotyping strategy for ARDS. Chest imaging is a component of the diagnostic criteria and is available on essentially all patients. The distinction between focal and diffuse attenuation can, seemingly, be made by most critical care providers and does not require costly send out labs. Yet, findings of the Lung Imaging for Ventilatory Settings in ARDS (LIVE) trial highlight potential challenges of this strategy. Patients were randomized to either the control arm (6 mL/kg predicted body weight ventilation, low PEEP, and FiO2 table) or the precision care intervention arm. In the intervention arm, patients with focal attenuation received higher tidal volumes and low PEEP, whereas patients with diffuse ARDS received 6 mL/kg tidal volumes and PEEP which was titrated to a plateau pressure of 30 cmH2O with the use of recruitment maneuvers after the titration. Prone positioning was mandatory for the focal group and usable only as a rescue in the diffuse group. Unfortunately, there was only moderate agreement between the local investigators and centralized adjudicators on whether the patients had focal or diffuse attenuation, and the personalized strategy seemed to improve survival only in correctly classified patients but not when patients were misclassified based on their chest radiograph [32]. These findings highlight the potential challenges of misclassification when identifying subphenotypes of ARDS.
Biological Subphenotypes
1. Protein biomarkers
Plasma protein biomarkers have been used to stratify disease severity and predict clinical outcomes in ARDS and may identify potential biological pathways that could be targeted by novel pharmacotherapeutic strategies. An exhaustive discussion of all the biomarkers used to subphenotype patients with ARDS is beyond the scope of this review, and readers are directed to recent summary reports on this topic [4,33,34]. Here, we highlight some of the notable biomarkers thought to be the most relevant in the diagnosis and prognostication of ARDS.
sRAGE is a biomarker of epithelial injury and alveolar fluid clearance which is known to be elevated in both early-onset and non-focal ARDS [22,30]. In a secondary analysis of the ARMA low tidal volume trial, baseline plasma RAGE levels were higher in patients with more severe ARDS and were higher in patients who died compared to survivors of ARDS. Further analysis revealed an interaction between RAGE levels and patient ventilation strategy, such that RAGE predicted mortality in patients who received high tidal volume ventilation but not low tidal volume ventilation [35]. Von Willebrand factor (vWF) antigen has also been heavily studied in ARDS. Higher levels of vWF are indicative of endothelial activation and endothelial damage. vWF levels are elevated in indirect lung injury and are a predictive biomarker for the development of ARDS [17,36,37]. Elevated levels of vWF are associated with mortality, prolonged duration of mechanical ventilation, and prolonged and refractory hypoxemia [38]. Tumor necrosis factor-α (TNF-α) is a pleotropic cytokine which has been linked to inflammation, endothelial permeability in the lungs, and impaired alveolar fluid clearance. TNF-α levels are significantly higher in patients with ARDS when compared to patients with severe pneumonia or controls [39]. TNF-α binds to and activates TNF-receptor 1 (TNFr1) which, in turn, activates downstream pro-inflammatory signaling, apoptosis, and impaired alveolar fluid clearance [40]. Work by Parsons et al. [41] suggests that TNFr1, but not TNF-α, have prognostic value in patients with ARDS. Interleukin 6 (IL-6) is another inflammatory cytokine mechanistically implicated in ARDS and a pharmacotherapeutic target specifically in COVID-19 ARDS. Interestingly, values of IL-6 are 10 to 200-fold higher in “hyperinflammatory” ARDS when compared to severe COVID-19 ARDS [42]. Ang-2, another marker and mediator of vascular endothelial activation, is elevated in patients with both ARDS and sepsis and is associated with severity of illness in sepsis, development of ARDS in sepsis, and higher mortality [43]. Other plasma biomarkers consistently associated with ARDS mortality include IL-8, intracellular adhesion molecule 1, thrombomodulin, plasminogen activator inhibitor 1 (PAI-1), and protein C (lower).
Despite this wealth of information, the use of any single protein biomarker to subphenotype patients with ARDS is likely to oversimplify complex inflammatory cascades which have networks of upstream activators and downstream targets. Modification of any one of these biomarkers may be advantageous in terms of reducing the aberrant inflammatory response seen in ARDS but may simultaneously abrogate the host’s necessary response to active infection. Furthermore, it is unclear whether a given biomarker associated with poor prognosis is causally linked to that poor outcome, or elevated to compensate for other pathways that are actually driving the poor outcome, or in fact is related to another confounding variable. More robust strategies may include aggregating mechanistically related biomarkers to establish subphenotypes of endothelial injury (i.e., Ang-2, vWF), epithelial injury (i.e., surfactant protein-D, club cell secretory protein, sRAGE), or systemic inflammation (i.e., IL-6, IL-8, TNFr1) [44], and integration with experimental models of lung injury and sepsis will likely be required to conclusively establish mechanisms.
2. Transcriptomics
Sweeney et al. [45] attempted to identify a transcriptomic signature associated with ARDS using multicohort analysis of gene expression in whole-blood but found that unique gene expression in ARDS patients was significantly confounded by overall inflammation and the presence of sepsis. Seven genes were identified that were best able to separate ARDS from non-ARDS, yet this model performed poorly in cross validation [45]. More work is needed to determine whether transcriptomic signatures differ between more defined subgroups of patients with ARDS (i.e., direct vs. indirect; early vs. late), and whether transcriptomic phenotypes identified in sepsis [45-50] can be observed in ARDS cohorts, to establish whether transcriptomic subphenotyping is a future possibility.
Unbiased Approaches to ARDS Subphenotypes
Lacking any consensus as to whether etiology, timing, nature of insult, physiology, or biological biomarkers are the best way to combat heterogeneity in ARDS, numerous investigators have sought to use unsupervised clustering methodologies to identify novel subphenotypes of ARDS. One approach used LCA to combine available clinical and biological data from National Heart, Lung, and Blood Institute (NHLBI) clinical trials to identify two novel subphenotypes of ARDS, termed “hyperinflammatory” and “hypoinflammatory” [51-53]. The “hyperinflammatory” group is characterized by higher plasma levels of IL-6, IL-8, and TNFr1 and lower plasma protein C and serum bicarbonate. These subphenotypes now have been replicated in at least seven different clinical cohorts [53-56]. Reliably, the hyperinflammatory group has appreciably higher mortality and worse clinical outcomes than the “hypoinflammatory” group [53]. Furthermore, in secondary analysis of completed trials, the subphenotypes appear to respond differently to PEEP, fluid management, and simvastatin [53,54,56]. Drohan et al. [57] utilized a similar LCA-based strategy in a cohort of patients with acute respiratory failure, not necessarily meeting the Berlin definition for ARDS. Their analysis included 22 clinical and molecular biomarkers and identified two groups of patients, also termed “hyperinflammatory” and “hypoinflammatory.” The “hyperinflammatory” group was defined by higher levels of leukocytes, creatinine, inflammatory biomarkers and lower serum bicarbonate. The hyperinflammatory group had significantly worse 30- and 90-day survival and fewer ventilator free days [57]. Bos et al. [58] used an alternative unsupervised classification strategy, cluster analysis, to identify two distinct clusters of patients using solely plasma biomarkers of endothelial activation, inflammation, and coagulation, independent of patient outcomes. These clusters were labeled “uninflamed” and “reactive.” Similar to the previously described “hyperinflammatory” subphenotype, the “reactive” subphenotype had a higher mortality and more multiorgan failure compared to the uninflamed cohort [58].
These unsupervised subgroup assignments were generated using various combination of plasma protein biomarkers plus/minus clinical data, making real-time implementation challenging. To circumvent this challenge, Sinha et al. [59] capitalized on machine learning algorithms and regression modeling to develop a parsimonious model for ARDS subphenotype identification. Three (IL-8, bicarbonate, and protein C) or four variables (IL-8, bicarbonate, protein C, vasopressors) accurately classified patients to the hypo or hyperinflammatory subphenotypes with a strong positive correlation with assignment based on LCA [59]. Similarly, Bos et al. [58] found that a limited set of biomarkers combining IL-6, interferon γ, ANG1/2, and PAI-1 could effectively differentiate the “uninflamed” and “reactive” subphenotypes with a high area under the ROC. The Kitsios group, too, developed a more parsimonious model by which to assign subphenotypes using bicarbonate, TNFR-1, Ang2, and procalcitonin [57].
Despite these advances, there are still limitations in the use of this technique as most of these protein biomarkers are not clinically available. Sinha et al. [60] went on to attempt to sidestep the need for protein biomarkers by using machine learning to identify and validate models which use only readily available clinical variables to assign patients to inflammatory ARDS subphenotypes. In a separate validation cohort, the area under the curve (AUC) of this clinical classifier model was 0.95, with a strong positive correlation between class assignment by LCA and the clinical classifier model. This anthology of work supports that unsupervised clustering techniques using a combination of clinical and protein biomarkers have identified two distinct subphenotypes of patients with ARDS with dramatically different mortalities and a differential response to previously trialed ARDS therapies.
Most recently, Chotalia et al. [61] have utilized LCA to identify novel cardiovascular subphenotypes in ARDS. They combined transthoracic echo and clinical variables from more than 1,000 patients who were diagnosed with ARDS. They identified three distinct cardiac subphenotypes differentiated by the degree of right ventricular (RV) dysfunction. The “RV dysfunction” subphenotype is characterized by RV dilation but with preserved RV function, has a mortality of approximately 40%, and is not independently associated with mortality. The “RV failure” subphenotype is characterized by RV dilation and impaired RV function, portends a mortality of greater than 70%, and is independently associated with mortality with an odds ratio of 6.9. The “hyperdynamic” subphenotype has a high cardiac output, low systemic vascular resistance, and is associated with a systemic hyperinflammatory state. This hyperdynamic subphenotype has a 59% mortality and does independently predict an increased risk of death. Chotalia et al. [61] established a parsimonious model to divide patients into these distinct subphenotypes using parameters available by bedside transthoracic echocardiogram and found that measures of RV systolic dysfunction and cardiac index can accurately discriminate the subphenotypes.
Subphenotyping Implications
1. Clinical trials
One potential benefit of subphenotyping patients is to enrich clinical trials. The current strategy of post hoc analysis of completed clinical trials which involve retrospective subphenotype assignment and evaluation of differential treatment effect has advanced the field but should not be practice changing. Subgroup analysis often yields underpowered or inaccurate results. Under ideal circumstances, we would enroll patients in clinical trials who are most likely to have the outcome of interest (prognostic enrichment) and who are most apt to benefit from the proposed intervention (predictive enrichment). These strategies were used in prognostically enriched by ARDS severity (Prone Positioning in Acute Respiratory Distress Syndrome trial [PROSEVA]) and in the Randomized Evaluation of COVID-19 Therapy trial (RECOVERY) trial of tocilizumab (predictively enriched by elevated C-reactive protein and hypoxemia), respectively [62,63]. At the same time, enrichment strategies risk limiting the generalizability of a study’s findings. Furthermore, for these strategies to be pragmatically possible, subphenotyping must be complete prior to patient randomization, which necessitates rapid, reliable, easy to interpret, point-of-care testing strategies. Fortunately, such strategies are currently under development and validation (NCT04009330).
As promising as these approaches seem, the recombinant human interleukin-1 receptor antagonist (rhIL1RA) Sepsis Syndrome Study should serve as a cautionary tale [32,64]. In vitro and animal model data suggested that rhIL1RA would bind the interleukin-1 receptor, competing with interleukin-α and -β, reducing vascular permeability and inflammatory cytokine production. Yet, clinical trials targeting this pathway repeatedly failed to demonstrate clinical benefit [64,65]. Furthermore, a retrospective analysis revealed that in patients with lower baseline levels of IL1RA, administration of supplementary rhIL1RA was associated with paradoxically increased patient mortality, while patients with higher endogenous IL1RA levels seemed to benefit from rhIL1RA [66]. This somewhat surprising finding highlights the complexity of the biological systems being targeted and our superficial understanding of mechanism. A priori stratification should ensure that both presumed responders and non-responders are enrolled in prospective clinical trials to confirm differential treatment response by biomarker stratification and ensure that no inadvertent and unexpected harm is caused, unless there is very convincing prior evidence of benefit only in one subgroup and/or harm in another.
Adaptive clinical trials with or without the integration of point-of-care biomarker platforms offer great promise in ARDS. Such designs could involve determination of a patient’s biological or molecular subphenotype a priori, stratified randomization, and subsequent adaptation based on the performance of individual interventions in patients with a particular subphenotype. As an example, all patients with ARDS could be enrolled and randomized in a trial of tocilizumab with real-time quantification of a biomarker such as IL-6. If interim analysis suggested a differential response to tocilizumab based on the levels of IL-6, then the biomarker-negative group could be dropped and further enrollment could focus on the biomarker-positive group [67].
Future Directions
Studies are needed to prospectively validate the subphenotypes that been identified using retrospective techniques. These studies should a priori define phenotypes but in most cases should enroll both suspected responders and non-responders to establish definitively what subphenotypes are non-responders. Research and development on new pharmacotherapeutic options for ARDS should persevere while U.S. Food and Drug Administration approved drugs with mechanistic plausibility or that have demonstrated benefit in conditions with biological overlap (i.e., sepsis) should be considered for repurposing. Simvastatin is an excellent example of an already approved drug which warrants prospective exploration based on the hypothesis-generating data indicating a heterogeneity of treatment effect of simvastatin based on the inflammatory subphenotype [56]. However, this essential clinical trial cannot be conducted unless the challenge of point-of-care phenotyping is overcome.
New research is focusing on whether subphenotypes of ARDS transcend individual critical illness syndrome. Heijnen et al. [68] applied clustering methods previously used to define the “reactive” and “uninflamed” subphenotype in ARDS to critically ill patients without ARDS and identified two distinct patient populations with similar odds of mortality, prolonged duration of mechanical ventilation, and blood leukocyte gene expression. Gene expression profiles clustered more by subphenotype then by syndromic diagnosis, alluding to shared mechanisms across critical illness syndromes [68]. Such work could move the field in a direction of enrolling patients with a treatable trait rather than with a diagnosis of a loosely defined syndrome. We also need more studies of the trajectories of subphenotype assignment over time. Most subphenotype assignments are based on enrollment data. In rare cases, subphenotype data extends out to hospital day three or four [16,69]. Metabolomics, genomics, transcriptomics, proteomic, and microbiome studies are underway in ARDS and will provide more insight into disease mechanism and outcome. Eventually, multi-omics may allow for deeper subphenotyping in ARDS and a more comprehensive understanding of the patient’s interaction with their environment.
Conclusion
The current standard of care for patients with ARDS remains supportive. Inherent heterogeneity in ARDS has slowed identification of effective therapeutic strategies. Numerous potential approaches to subphenotyping have been described, which suggest that a precision medicine approach in ARDS may be effective, but ongoing work is needed to develop rapid point-of-care subphenotyping, to advance our understanding of biologic differences between identified subphenotypes, and for prospective validation of currently proposed ARDS subphenotypes.
Notes
Authors’ Contributions
Writing - original draft preparation: all authors. Writing - review and editing: all authors. Approval of final manuscript: all authors.
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Funding
No funding to declare.