Introduction
One-third of the world's population is infected with Mycobacterium tuberculosis. In high tuberculosis (TB) burden countries, the rapid diagnosis and treatment of infectious TB are major concerns. However, in low TB burden countries, the main TB control policy is to screen for active and latent TB in selected risk groups, including immigrants from TB-endemic areas.
The TB burden in South Korea is intermediate, with an annual incidence of 80/100,000 in 20151. To achieve the Sustainable Development Goals for 2030 proposed by the World Health Organization, the Korean government launched a comprehensive plan for TB control in 20132, and it is scaling up the policy to include intensive TB screening for immigrants from TB endemic areas because the number of immigrants with TB has increased each year in Korea3.
The most common race of TB patients among immigrants is Korean-Chinese (Chosun race) from northeastern China, including the Yanbian Korean Autonomous Prefecture4. However, it is essential to determine the molecular epidemiology of Mycobacterium tuberculosis isolates and identify the transmission dynamics in the Korean-Chinese population to define a strategy for immigration TB screening. A clustering of M. tuberculosis strains from native Koreans (K family) was reported5, and it was different from that of foreign-born patients according to restriction fragment length polymorphism profile analyses6.
Recently, genome organization studies have become possible because whole genome sequences, including the genomes of microorganisms, can be sequenced using next-generation sequencing (NGS)7. Moreover, phylogenetic relationship studies using the Multiple Alignment of Conserved Genomic Sequence with Rearrangements (Mauve) package based on genomic bioinformatics are promising, because Mauve package is suitable for sequence comparison not posed by short sequences in the presence of rearrangements and horizontal transfer8.
The aims of this study are to characterize TB strains isolated from immigrants from China based on molecular and bioinformatics as well as to compare them with clinical isolates from native Chinese and Korean individuals.
Materials and methods
1. Bacterial culture conditions and DNA extraction
Two new clinical isolates from immigrants who were treated in Korea University Guro Hospital and Korea University Ansan Hospital were used for whole genome sequencing. The mycobacterial cultures were grown at 37Ōäā on L├Čwenstein-Jensen medium. Strains harvested at the early exponential phase were used for DNA extraction. All DNA extraction was performed as previously described9. At least one loop-full of cells (100 mg wet weight) were washed twice with TE (Tris-HCl pH 8.0, 10 mM; EDTA 1 mM) and an equal volume of saturated cesium chloride solution containing 1% Triton-X was added to it. Further purification was performed by the usual phenol-chloroform extraction and DNA was spooled out after adding 0.5 vol. of 5 M ammonium acetate and 0.75 vol. of isopropanol; it was washed with alcohol, partially dried and suspended in TE buffer. It was then treated with RNase A (50 ┬Ąg/mL) for 30 minutes at 37Ōäā, re-extracted with chloroformisoamyl alcohol, precipitated and finally re-suspended in TE buffer.
2. M. tuberculosis genome sequences assembly
Whole-genome shotgun sequencing of the two strains was carried out using PacBio SMRT sequencing technology10 with ~150├Ś depth. For assembly of the genomes, we applied the recently described hierarchical genome assembly process (HGAP) to the SMRT cells of sequencing data generated from an 8- to 10-kb insert library11. Pre-assembly error correction was performed with HGAP of SMRT analysis version 2.3.0 (Pacific Biosciences, Menlo Park, CA, USA) using default parameters. Error-corrected reads were then assembled using Celera Assembler12. To produce circular full-length sequence, each end section was resolved manually.
3. Bioinformatics study and Mauve alignments for M. tuberculosis strains
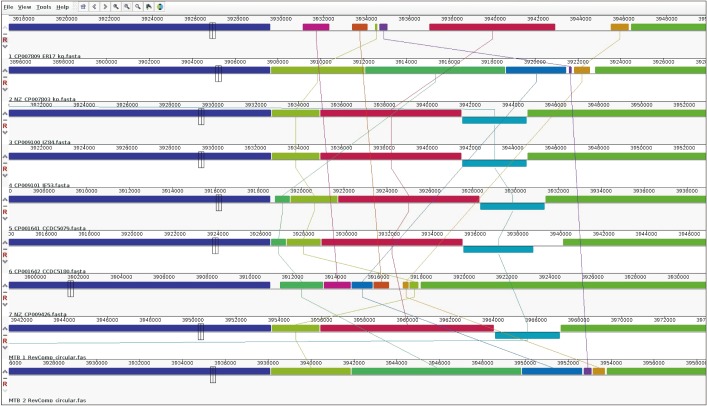
We compared the genome topology network (GTN) of each strain using a bootstrap topology tree and detected the locally collinear blocks of the conserved segments using a modified MUSCLE (multiple sequence alignment method with reduced time and space complexity) global alignment algorithm. We used the Mauve package for the identification and alignment of the conserved genomic DNA sequences of the nine M. tuberculosis strains8. Initially, local multiple alignments that had unique subsequences shared by two or more input genomes were selected, and ungapped extension was performed until the seed pattern no longer matched. Then, a progressive genome alignment according to a guide tree was built up after computing a pairwise genome content distance using the neighbor joining method and a pairwise breakpoint distance matrix. During progressive alignment, the breakpoint penalty according to the expected level of sequence divergence and the number of well-supported genomic rearrangements among the pair of input genomes were scaled. Anchor alignment using a global genome alignment algorithm was performed, and alignments that had unrelated sequences were ultimately rejected. The algorithm began with the initial set of matching regions (multiple maximal unique matches) represented as connected blocks. The matches were partitioned into a minimum set of collinear blocks. Each sequence of identically colored blocks represents a collinear set of matching regions. One connecting line is drawn per collinear block.
This study was approved by the Institutional Review Board (IRB) of Korea University Ansan Hospital (KUAS15157-001) and Korea University Guro Hospital (2017GR0301) with waivers of informed consents.
Results
1. Whole-genome sequences of clinical isolates of two immigrants
Table 1 presents the characteristics of two complete whole genome sequences from immigrants. The complete genome sequence of M. tuberculosis MTB_1 has been assigned the GenBank accession number CP020381.2. The M. tuberculosis MTB1 genome comprises one chromosome of 4,433,542 bp. In total, 4,306 genes comprising 4,255 coding sequences (CDS), three rRNA genes (5S, 16S, and 23S), 45 tRNAs, and three non-coding RNAs (ncRNAs) were annotated using NCBI Prokaryotic Genome Annotation Pipeline (PGAP; http://www.ncbi.nlm.nih.gov/genome/annotation_prok). The patient with the MTB_1 strain was a 39-year-old woman. She had recently emigrated from Shandong Province in China to South Korea to marry a Korean. Her chest computed tomogram showed a cavity in the left lung apex representing the reactivation of a previous TB infection.
The complete genome sequence of M. tuberculosis MTB_2 has been assigned the GenBank accession number CP022014.1. The M. tuberculosis MTB2 genome comprises one chromosome of 4,417,716 bp. In total, 4,290 genes comprising 4,239 CDS, 117 pseudo genes, three rRNA genes (5S, 16S, and 23S), 45 tRNAs, and three ncRNAs were annotated. The patient with MTB_2 strain was a 33-year-old man. He had moved to South Korea from the Yanbian Korean autonomous prefecture where most Korean-Chinese (Chosun race) reside, 2 years before the TB diagnosis was confirmed. His chest computed tomogram showed centrilobular nodules and branching opacities in both upper lung fields.
2. Epidemiologic characteristics of TB strains for comparison
As shown in Table 2, the genomic sequences of seven strains (from Nos. 1 to 7) from native Chinese and South Korean individuals, which were obtained from a public website, were compared with MTB_1 and MTB_2 from the immigrants to South Korea. The Nos. 1 and 2 strains, which are known to be strains from native South Koreans, were isolated during an outbreak of pulmonary TB in high schools13,14. The Nos. 3, 4, 5, and 6 strains were reported as strains from native China. The Nos. 3 and 4 strains were isolated from Chinese individuals living in Zunyi, which is located in southwest China15; the Nos. 5 and 6 strains were isolated from Chinese individuals living in Fujian Province, which is located in southeast China16; and the No. 7 strain was isolated from Chinese individuals living in Beijing, which is located in northwest China17. When the strains from the immigrants were analyzed, the MTB_2 strain was similar to the No. 1 and No. 2 strains isolated from native Koreans. On the other hand, the MTB_1 strain was similar to the strains isolated from the native Chinese individuals.
3. Mauve alignment and phylogenetic trees of nine M. tuberculosis strains
Figure 1 represents the Mauve alignment for nine M. tuberculosis strains, which shows collinear set of matched colored regions. And, in Figure 2, the phylogenetic trees exhibit similarities and differences in topology. The phylogenetic analysis of the GTN resulted in a rooted phylogenetic tree grouping the nine strains into two lineages: (1) strains from Chinese individuals and (2) strains from Korean individuals. However, even though the MTB_1 strain was grouped to a lineage from Chinese individuals (from the Nos. 3 to 7 strains), the MTB_1 strain was more similar to the Nos. 3 and 4 strains (Zunyi, China) than to the Nos. 5, 6, and 7 strains (Fujian Province and Beijing, China). On contrast, MTB_2 was grouped to a lineage from the Korean individuals (Nos. 1 and 2) that was different from the strains from the Chinese individuals, closer to the No. 2 strain than to the No. 1 strain.
Discussion
The whole-genome sequences of two clinical isolates from the immigrants were deposited in the GenBank under accession numbers CP020381.2 and CP022014.1. Furthermore, after classifying the alignments of the conserved genomic DNA sequences from nine strains into two lineages using phylogenetic trees based on Mauve alignments, we were able to infer the origin of the strains from the immigrants.
Considering the similarities of the MTB_2 strain isolated from a male Korean-Chinese (Chosun race) individual who emigrated from Yanbian Province, China, with the native Korean K1 strains (Nos. 1 and 2), he may have been infected with a strain from native Koreans after immigration, as Jeon et al.6 reported, because the Korean K1 strains have been reported as the most widely distributed characteristic strains in South Korea5. Otherwise, another plausible explanation is that the strains from Yanbian Province, where Chosun race live because of the geographical proximity to the Korean Peninsula, may have the same lineage as the native Korean K strains. Therefore, additional molecular epidemiology studies with large numbers of samples must be conducted to characterize the strains from Korean-Chinese (Chosun race) individuals.
However, considering the similarities of the MTB_1 strain isolated from a Chinese woman who emigrated from Shandong Province in China with the native Chinese strains (Nos. 3, 4, 5, 6, and 7), she is thought to have settled in South Korea without a detailed proper screening for active or latent TB during the immigration process. Therefore, to prevent the inflow of TB by immigrants, a more aggressive and thorough TB screening process for immigrants must be performed to reach the goals of the 2030 TB elimination project in South Korea.
Because the distribution of specific resistance-conferring mutations is fairly constant worldwide, suggesting that drug resistance has arisen via common mechanisms18, further bioinformatic analyses of multi-drug resistant TB or extensively drug-resistant TB isolates from China and Korea are needed, which should include a large number samples with accurate information including phenotypic resistance, to determine the phylogenetic relationship with drug resistance.
This study had several limitations. First, we could not analyze a sufficient number of isolates because it was very difficult to extract and isolate mycobacterial DNA from stock strains, as a time-consuming and tedious process was necessary to disrupt the thick and lipopolysaccharide-rich cell wall without causing damage to the genomic DNA, even though a recently approved method was used9. Second, it seems too early to consider NGS a generalized genome analysis method for obtaining full M. tuberculosis sequences, annotation refinement, and orthology assignment because it is very expensive. Finally, there have been no reports on the differences between the Chinese strains and the Korean strains using bio-informatics analyses; therefore, the phylogenetic trees for the strains constructed using the small number of samples were likely not definite.
In conclusion, phylogenetic trees based on Mauve alignments supposed to reveal the dynamics of TB transmission from immigrants to South Korea, providing important information for the scaling up of the TB screening policy for immigrants, especially from China.

 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Print
Print Download Citation
Download Citation