Introduction
DNA methylation at the CpG islands within the promoter regions of tumor suppressor genes (TSGs) and cancer-associated genes plays a critical role in human cancers1,2. Aberrant promoter methylation may affect the genes involved in cell cycle control (p16INK4Ap15, Rb, p14), DNA repair (MGMT, hMLH1), cell adhesion (H-cadherin, CDH-1), signal transduction (RASSF1A), apoptosis (DAPK, TMS1), and cell differentiation (RARβ2)3. Cyclin-dependent kinase inhibitor 2A (CDKN2A), retinoic acid receptor beta (RARβ), and Ras association domain family member 1A (RASSF1A) are important TSGs that are frequently found in lung cancer patients. Hypermethylation at the promoter 5'-CpG island of these TSGs is frequently associated with gene silencing and occurs during carcinogenesis.
Among the methods used for assessing methylation, methylation-specific polymerase chain reaction (MSP), which is performed after bisulfite modification, is the most widely used method. The main advantages of MSP are that it is a very sensitive method, permitting the analysis of small and heterogeneous samples; it can be used on paraffin-embedded samples; it is specific for relevant CpG sites; and it eliminates the requirement for restriction enzymes, thus preventing problems resulting from and associated with incomplete enzymatic digestion. However, MSP tends to be more of a qualitative method than a quantitative one. Therefore, pyrosequencing is ideally suited for the simultaneous analysis and quantification of the degree of methylation at multiple CpG islands within each gene promoter region. Pyrosequencing is based on the sequencing-by-synthesis principle and is a bioluminometric real-time DNA sequencing technique that uses a cascade of 4 enzymatic reactions, producing light upon nucleotide incorporation 4.
In this study, we analyzed methylation status at the CDKN2A, RARβ, and RASSF1A promoters in non-small cell lung carcinoma (NSCLC) tumor samples by using pyrosequencing. Then, we evaluated the association between methylation at the promoter regions of these TSGs and the clinicopathological parameters of the NSCLCs. In addition, we investigated the correlation between the hypermethylation of CDKN2A and the loss of p16INK4A immunoexpression.
Materials and Methods
1. Patients and clinicopathological parameters
We analyzed 53 NSCLC patients who underwent surgery from 2000 to 2007 at Konyang University Hospital. This study had local ethics committee approval obtained from the Chungnam National University Hospital's Institutional Review Board. We retrieved these formalin-fixed, paraffin-embedded tissue blocks for analyzing of promoter methylation and immunohistochemical staining. We also reviewed the medical records of the NSCLC patients. The patients were characterized by clinicopathologic parameters (Table 1). Histopathological classification was assessed according to the World Health Organization (WHO) criteria. Tumor-node-metastasis (TNM) staging followed the American Joint Committee on Cancer (AJCC) staging system as revised in 2010 (7th edition). Squamous cell carcinoma (SqCC), adenocarcinoma, undifferentiated large cell carcinoma (LCC), which are major histological types of NSCLC, accounted for 32 (60.4%), 17 (32.1%), and 4 (7.5%), respectively. TNM stage following surgical resection was stage I in 24 (45.3%), stage II in 10 (18.9%), stage III in 14 (26.4%) and stage IV in 5 (9.4%). The median length of follow-up after the operation was 33 months (range, 1.5~80 months). Within follow-up periods, 23 (43.4%) patients died of cancer-related causes.
2. DNA extraction and bisulfite modification
All 53 samples were obtained by dissection of tumor areas and 13 samples were obtained by dissection of normal areas apart from tumors. Deparaffinization was carried out by adding 1 mL xylene to the microtube containing paraffin-embedded tissue sections and shaking incubation at normal temperature. This step repeated two or three times then spun down at 11,000 rpm for 3 minutes. The supernatant was removed, two identical washes with 100% ethanol for 15 minutes, spinning at 11,000 rpm for 3 minutes, was followed by drying at 37℃ incubator for 15 minutes. DNA extraction was prepared using the QIAamp DNA Micro Kit (Qiagen, Hilden, Germany) as specified by the manufacturer's instructions.
Bisulfite treatment was performed to convert unmethylated cytosine to uracil and leaving methylated cytosine unchanged. Bisulfite treatment of 200 ng of each sample was subjected using the EZ DNA Methylation-Gold kit (Zymo Research, Orange, CA, USA) according to the manufacturer's instructions. The bisulfite converted DNA was eluted in 20 µL of elution buffer (Zymo Research). DNA samples were immediately stored at -20℃ until to use.
3. Primer design and pyrosequencing methylation analysis
Polymerase chain reaction (PCR) assays were designed to amplify a part of the CpG islands in the CDKN2A, RARβ, and RASSF1A genes. Primers were designed by using the PSQ assay design program (Biotage, Charlotte, NC, USA). The primer sequence of CDKN2A, RARβ, and RASSF1A genes was listed in the Table 2.
PCR reaction was carried out in a volume of 50 µL with 20 ng or less converted gDNA, 5 µL of 10× Taq buffer, 5 unit Hot/Start Taq polymerase (Enzynomics, Daejeon, Korea), 4 µL of each 2.5 mM dNTP mixture, 2 µL of 10 pmole/µL PCR primers. The amplification was carried out according to the general guidelines suggested by pyrosequencing: denaturating at 94℃ for 15 minutes, followed by 40 cycles at 94℃ for 30 seconds, at 58℃ for 40 seconds, at 72℃ for 300 seconds and a final extension at 72℃ for 10 minutes. Confirmation of PCR product quality was established on 1.5% agarose gel stained with ethidium bromide. Pyrosequencing was performed using the PSQ96MA System (Biotage) according to manufacturer's instructions including single strand binding protein (PyroGold reagents; Biotage).
4. Construction of tissue microarray and immunohistochemistry
All the hematoxylin and eosin stained slides for each case were reviewed, and the areas of interest were marked on each slide. The tissue microarrays (TMAs) were constructed using the 5 mm punch on the Bee-Cher arrayer (UNITMA, Seoul, Korea). The corresponding region was circled on the 'donor' paraffin block using a marker pen. The samples were then arrayed on a 'recipient' block. A total 53 samples were available for the array.
Four µm-thick sections of the paraffin-embedded TMAs were deparaffinized with xylene and rehydrated in a series of graded alcohol solutions and microwave-treated for 25 minutes in a citrate buffer (pH 6.0). The antigen was retrieved with 0.01 M citrate buffer (pH 6.0) by heating the sample in autoclave (CHS-ACCE-860; JW Pharmaceutical, Seoul, Korea) at a controlled final temperature of 121℃ for 15 minutes. Endogenous peroxidase activity was blocked using 0.3% hydrogen peroxide. Nonspecific binding sites were blocked by incubating in 10% normal goat serum diluted with phosphate-buffered saline (PBS). Tissue sections were then incubated with p16INK4A mouse mAb (1:200, Abcam, Cambridge, MA, USA) for 60 minutes at room temperature. The sections were visualized with avidin-biotin-peroxidase complex and tissue arrays were counterstained with Harris' hematoxylin. Human brain tissue served as positive internal control for p16INK4A staining. Negative controls were incubated with PBS instead of a primary antibody.
5. Statistical analysis
All statistical analyses were performed using SPSS version 18 (SPSS Inc., Chicago, IL, USA) for windows. Group comparisons of the clinical variables were performed using chi-square tests or linear by linear association analysis. Comparisons of means were accomplished by independent sample t-tests or one-way ANOVA. The difference was considered to be statistically significant if the two-sided p-value was <0.05. Survival curves were plotted using the Kaplan and Meier method and statistical significance was determined by the Breslow test.
Results
1. Relationship between CDKN2A hypermethylation and p16INK4A immunoexpression and relationship between these factors and the clinicopathological parameters of NSCLC
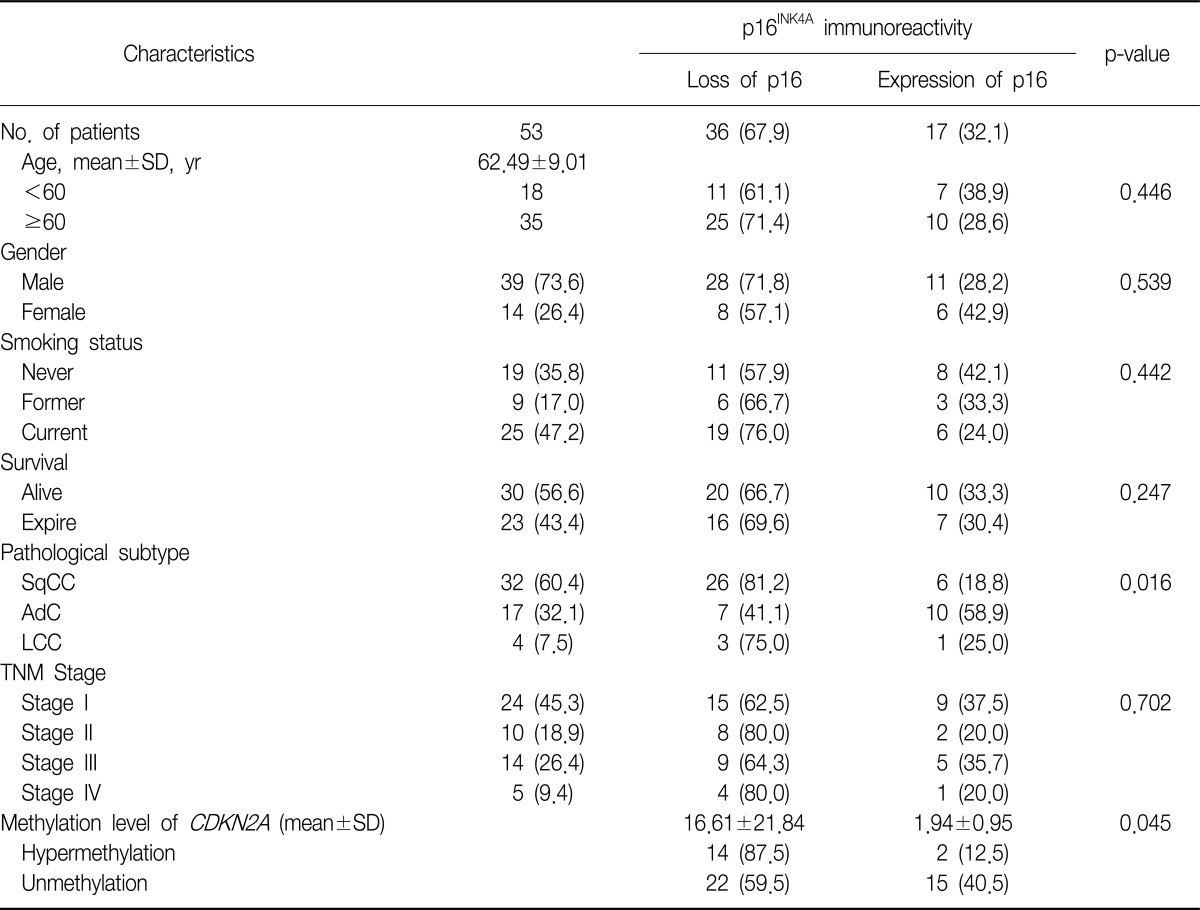
We examined the relationship between the CDKN2A methylation level and p16INK4A immunoexpression in 53 NSCLC tumor tissue samples and also examined the clinicopathological parameters of the NSCLCs (Table 1). Loss of p16INK4A immunoexpression was found in 36 samples (67.9%). Hypermethylation of CDKN2A was significantly associated with loss of p16INK4A immunoexpression (p=0.045). In this study, we showed a significant correlation between the CDKN2A gene hypermethylation status, which was identified by pyrosequencing, and the loss of p16INK4A immunoexpression, which was detected by immunohistochemical analysis, in the NSCLC tumor samples.
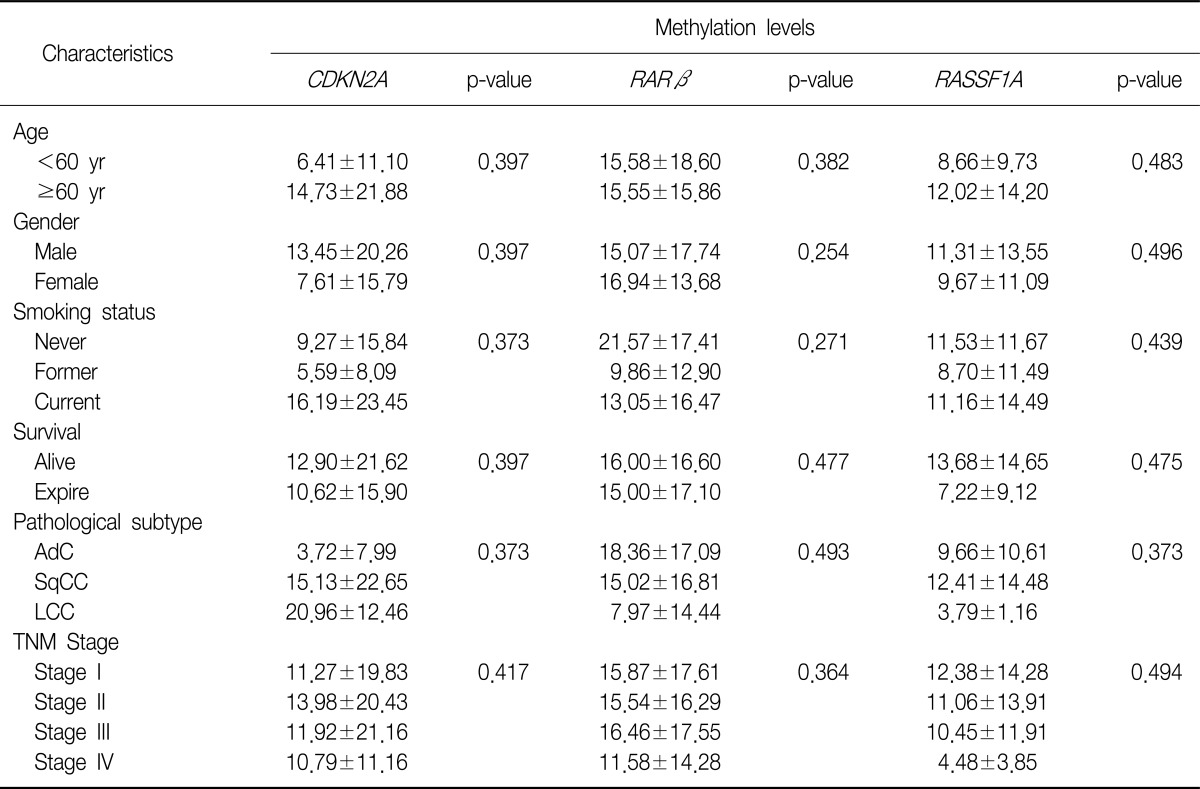
With regard to the clinicopathological characteristics of NSCLC, certain histopathological subtypes were found to be strongly associated with the loss of p16INK4A immunoexpression (p=0.016), i.e., SqCC and undifferentiated LCC showed more frequent loss of p16INK4A immunoexpression. However, no other clinicopathological parameter was associated with the loss of p16INK4A immunoexpression.
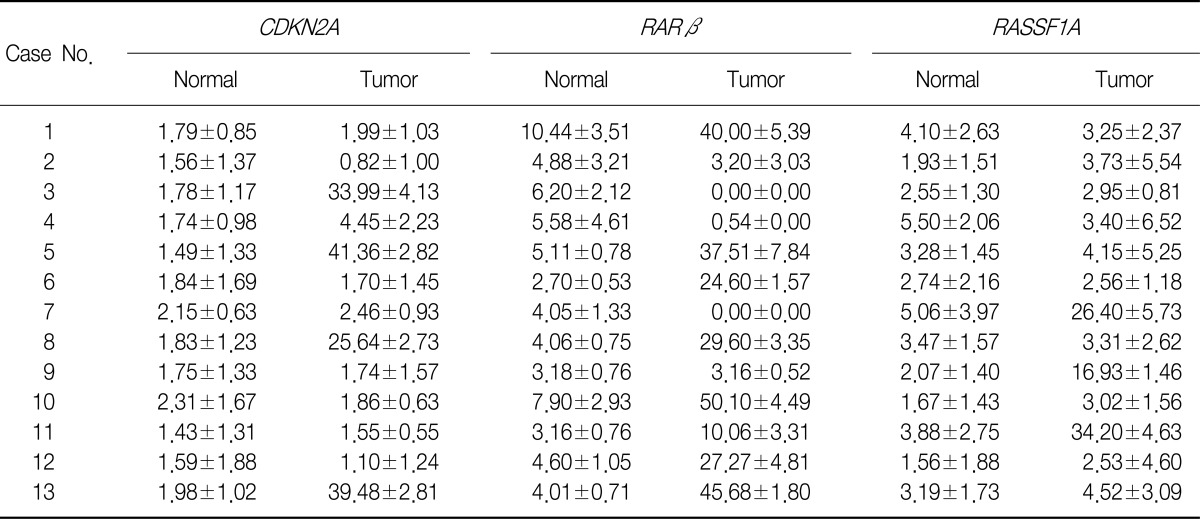
2. Methylation analysis of CDKN2A, RARβ, and RASSF1A promoters in tumor and normal lung tissues by using pyrosequencing
We evaluated the methylation status of the CDKN2A, RARβ, and RASSF1A gene promoters in 13 lung cancer tissue samples and 13 corresponding normal lung tissue samples by using pyrosequencing (Table 3, Figure 1). The mean methylation levels of normal lung tissues were 1.79, 5.07, and 3.15 and of tumor tissues were 12.16, 20.9, and 8.53 for CDKN2A, RARβ, and RASSF1A, respectively. On the basis of these results, we defined promoter hypermethylation in a tumor tissue as a 2-fold increase in normal tissue5, i.e., 3.58, 10.14, and 6.30 in CDKN2A, RARβ, and RASSF1A, respectively. We further analyzed an additional 40 tumor tissues by using pyrosequencing (Table 4).
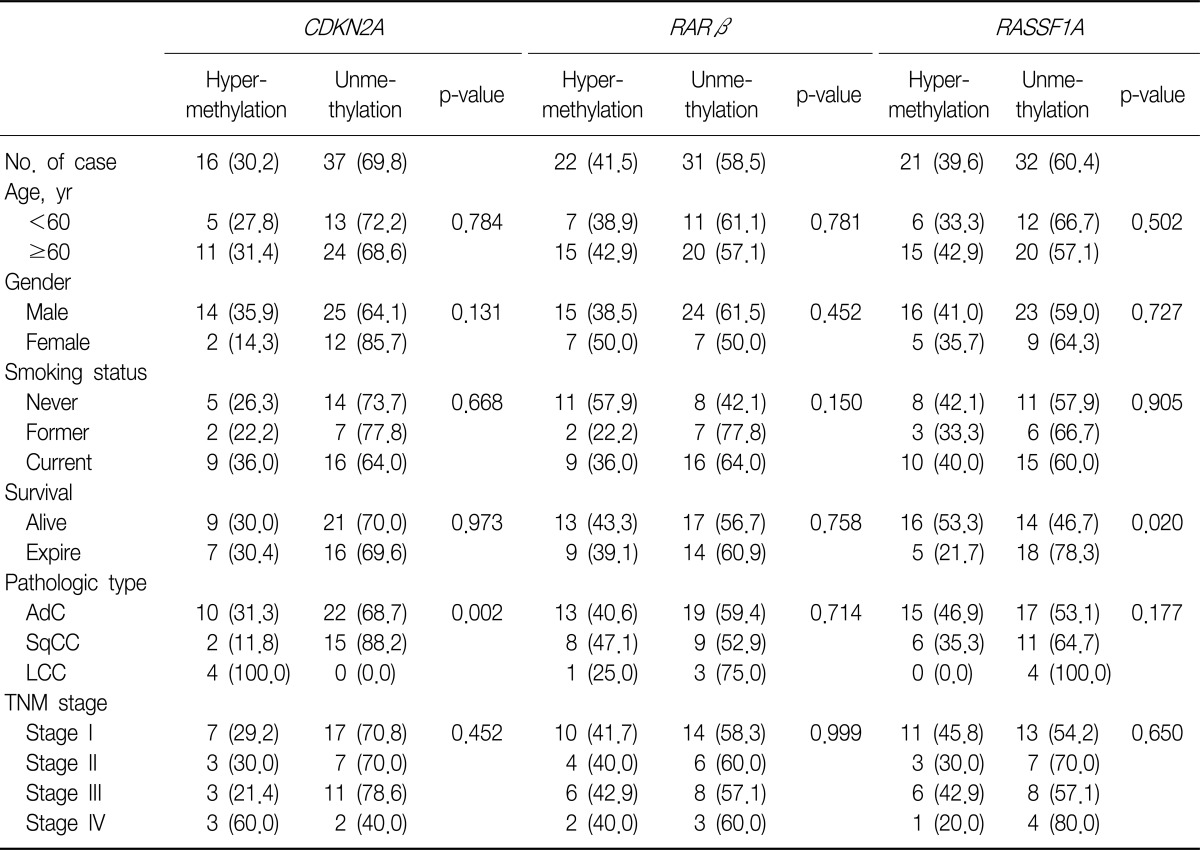
3. Methylation of CDKN2A, RARβ, and RASSF1A promoters and the correlation between their methylation status with the clinicopathological parameters of NSCLC
We evaluated the methylation status of the promoter regions of the CDKN2A, RARβ, and RASSF1A genes in 53 tumor tissue samples, as well as the correlation between methylation status of these promoter regions and the clinicopathological parameters of the samples (Tables 4, 5).
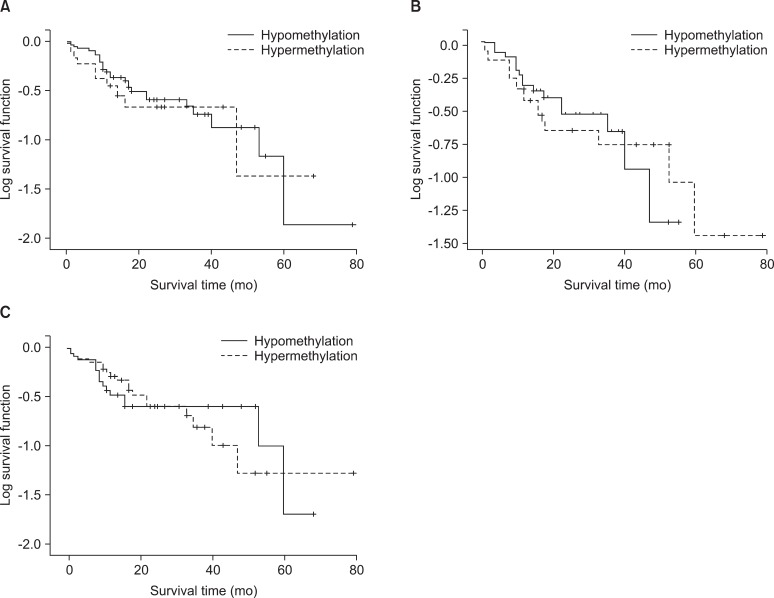
The incidence of hypermethylation at the CDKN2A promoter in the tumors was higher in the samples of undifferentiated LCC than in the samples of other subtypes (p=0.002) (Table 5). However, the number of LCC samples studied was too small to obtain meaningful results. The RARβ or RASSF1A gene promoters were not correlated with any clinicopathological parameters of NSCLC. We analyzed the effect of CDKN2A, RARβ or RASSF1A methylation on patient survival. Kaplan-Meier survival curves analysis showed that methylation level and patient survival were barely related (Figure 2).
Discussion
Lung cancer is the main cause of cancer-related deaths globally6. Lung cancer mainly comprises NSCLCs, which represent 85% of all lung cancer cases. Despite recent advances in early diagnosis and multimodality therapy, the prognosis for NSCLC is quite poor. Visbal et al.7 reported that the estimated 5-year survival rates were 15% in men and 19% in women. Therefore many studies have attempted to assess the specific molecular determinants that aid in detection of lung cancer at earlier stages and the mechanisms underlying tumor aggressiveness.
DNA hypermethylation refers to the addition of a methyl group to the cytosine ring of cytosines that precede a guanosine (CpG dinucleotides) resulting in the formation of methyl cytosine (5-methylcytosine). TSG contain CpG islands in their promoter regions, and many of them show evidence of methylation silencing8,9. In this study, we evaluated the methylation status of tumor suppression genes associated with lung cancer, such as CDKN2A, RARβ, and RASSF1A, by using pyrosequencing. Resultantly, we showed that the methylation level of the promoter regions of the aforementioned genes was much higher in the tumor tissues than in the adjacent normal tissues. Some researchers have shown that hypermethylation of the CpG islands of the specific promoter regions of specific genes such as RASSF1A, RUNX3, FHIT, and H-cadherin was closely associated with poor prognostic factors of NSCLCs, such as advanced stage, lymph node metastasis, and adverse survival10-13. In this study, CDKN2A hypermethylation was associated with the histopathological subtype of NSCLCs, i.e., undifferentiated LCC, although other clinicopathological characteristics were not related with the methylation status of CDKN2A, RARβ, and RASSF1A.
We also evaluated the immunoexpression of p16INK4A in NSCLC samples. SqCC and undifferentiated LCC showed immunoexpression loss of p16INK4A more frequently than other NSCLC subtypes. Many studies showed that CDKN2A gene hypermethylation was associated with loss of p16INK4A expression, wherease others showed that hypermethylation of the CDKN2A was related with overexpressed p16INK4A
1,14-16. In this study, we demonstrated that hyperrmethylation of the CDKN2A was significantly associated with loss p16INK4A immunoexpression in NSCLCs.
Many efforts have been made to develop effective methods for detection of DNA methylation. MSP has been found to provide useful results, mainly owing to its high sensitivity. However, MSP has several disadvantages: 1) This method yields qualitative rather than quantitative data; 2) It usually evaluates only a few CpG sites at the 3' end of the primers; 3) It lacks an internal control for evaluating the adequacy of bisulfite treatment, thus complicating the identification of false-positives results; 4) MSP is potentially susceptible to oversensitivity after numerous PCR cycles. In an attempt to overcome these disadvantages, we used pyrosequencing to determine the methylation status of the CDKN2A, RARβ, and RASSF1A promoters in NSCLC tumor samples. The advantages of pyrosequencing are that it is a highly reliable and quantitative sequencing-by-synthesis method with built-in internal controls for evaluating the adequacy of bisulfite treatment and can be used for analyzing DNA methylation at multiple CpG sites17-19. Pyrosequencing analysis can provide reproducible measurements of the average methylation levels in sequential CpG sites and can directly use PCR products, this method is rapid and accurate and can be used for simultaneous analysis of many samples. It is therefore well suited, for example, to clinical research applications where the methylation levels of several individuals need to be measured accurately over time to analyze the correlation with a disease status or response to treatment. However, owing to the limitation of pyrosequencing, i.e., sensitivity due to background noise20, the application of this method to clinical samples such as plasma and saliva may have little value21,22.
In conclusion, we quantitatively analyzed promoter methylation status by using pyrosequencing. We found that CDKN2A was hypermethylated in 30.2%, RARβ in 41.5%, and RASSF1A in 39.6% of the NSCLC samples. We showed a significant correlation between CDKN2A hypermethylation and p16INK4A immunoexpression loss. Despite the theoretical advantages of pyrosequencing, no correlation was found between promoter methylation status and survival rates.


 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Print
Print Download Citation
Download Citation