An Overview of Genetic Information of Latent Mycobacterium tuberculosis
Article information

Abstract
Mycobacterium tuberculosis has infected more than two billion individuals worldwide, of whom 5%–10% have clinically active disease and 90%–95% remain in the latent stage with a reservoir of viable bacteria in the macrophages for extended periods of time. The tubercle bacilli at this stage are usually called dormant, non-viable, and/or non-culturable microorganisms. The patients with latent bacilli will not have clinical pictures and are not infectious. The infections in about 2%–23% of the patients with latent status become reactivated for various reasons such as cancer, human immunodeficiency virus infection, diabetes, and/or aging. Many studies have examined the mechanisms involved in the latent state of Mycobacterium and showed that latency modified the expression of many genes. Therefore, several mechanisms will change in this bacterium. Hence, this study aimed to briefly examine the genes involved in the latent state as well as the changes that are caused by Mycobacterium tuberculosis. The study also evaluated the relationship between the functions of these genes.
Introduction
Mycobacterium tuberculosis (MTB) which is at least as old as human life on Earth, is one of the major challenges to human health [1,2]. Infection with MTB has been estimated in approximately one third of the global population [3]. The World Health Organization (WHO) reported that MTB is a preeminent cause of mortality (1.8 million deaths) worldwide in 2018, which is responsible for more deaths than human immunodeficiency virus and malaria [4]. Tuberculosis (TB)-infected persons are classified into two groups as (1) those with active TB presenting clinical or radiological manifestations of infection supported by laboratory evidence, (2) and those with latent tuberculosis infection (LTBI), which is an asymptomatic clinical presentation considering the largest reservoir for potential transmission [5]. Approximately 23% (1.7 billion people) of the world’s overall population suffers from LTBI [3].
The term LTBI was first proposed by Von Pirquet (1907), the ‘Godfather’ of the tuberculin skin test when he detected tuberculin skin reactions of ≥5 mm in children who did not manifest tuberculosis [6]. Lateral McCune et al. (1956‒1996) showed the persistence of latent tubercle bacilli for extended times after chemotherapy, shortly after the launch of isoniazid. Their well-designed investigation, named the ‘Cornell model’ by the academic institute wherein the research was conducted, can be regarded a main work on mycobacterial latency and its relation to tuberculosis chemotherapy [7]. The abundant contingent indications from consideration of the natural history of tuberculosis in humans and experimental animals indicate that MTB is able to adapt to prolong periods of dormant status in tissues, and that such dormant bacilli are the causative agent for latency of the disease itself. Besides, the dormant bacilli can resist eradication by antimycobacterial agents [8]. Accordingly, the WHO end TB Strategy targets treatment of diseased people as well as LTBI cases who are at the risk of advancement to TB disease. Therefore, prevention of LTBI development to active TB seems to be an essential public health objective, which could markedly decline the TB reservoirs [9].
The risk of TB recurrence during the life of an individual with confirmed LTBI is predicted to be 5%–15%, and most of them develop active TB during the first 5 years following early infection [10]. Nonetheless, the probability of LTBI development to TB disease is dependent upon various factors including bacterial, host, and environmental parameters. Basically, LTBI can be remarked as an equilibrium condition between host and mycobacteria [4]. In response to MTB infection, most patients represent a robust immune response, resulting a low bacillary burden in the absence of clinical and radiographic findings. In this situation, the host immune response prevents developing active disease, and the tubercle bacilli evade immune elimination [11].
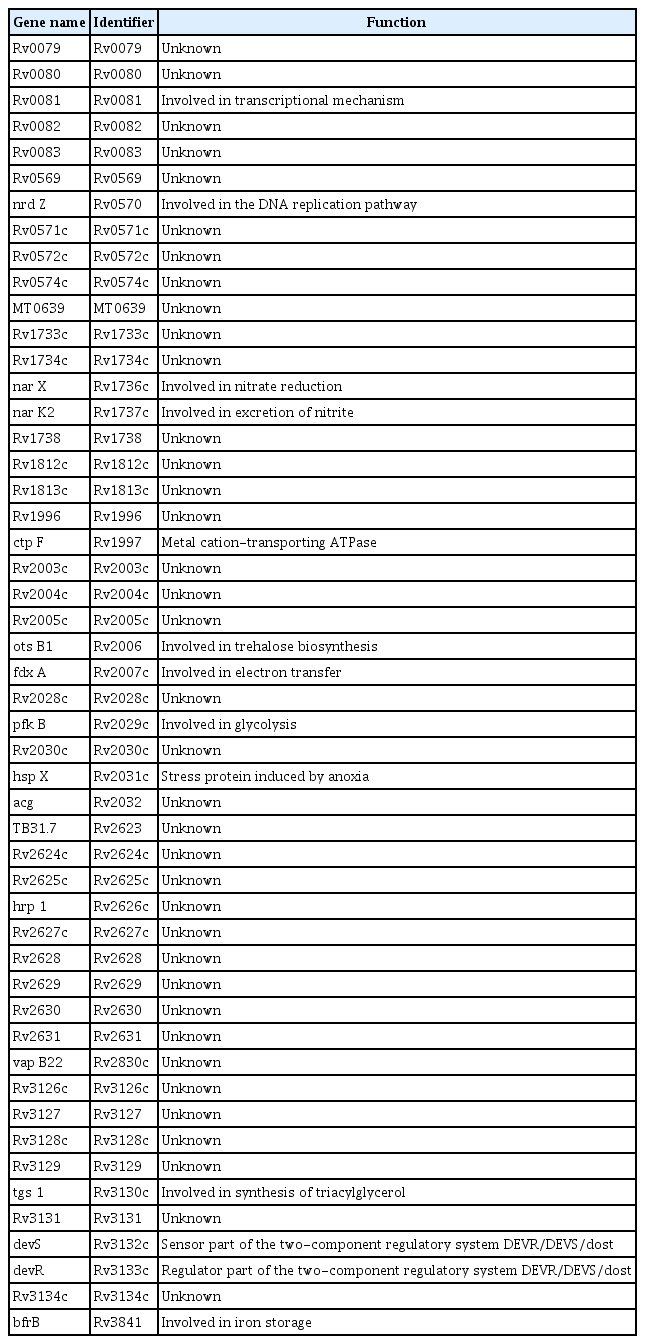
In LTBI stage, bacilli can survive in a modified physiological state in MTB-infected macrophages and escape to activity immune system [12]. However, despite several years of examination, the accurate locality of the latent mycobacteria is still mysterious. Moreover, in some cases of LTBI, persistent bacilli appear as the antibiotic-tolerant organisms which are nonreplicating and exhibit reduced metabolic activity. Reduced susceptibility to elimination by cell wall structure inhibitor agents such as isoniazid is a general characteristic of persistent bacilli in LTBI [13]. The structural and physiological changes which occur permanently in MTB are associated with the expression of various genes, enabling the bacteria to survive under latency conditions [14]. Therefore, for a better understanding of latency in MTB, the genes allowing bacteria to survive and escape from the immune system need to be more investigated [15]. In this regard, we tried to overview the genetic information of latency in MTB [16]. The general characteristics of the genes examined in this review are summarized in Table 1. The genes investigated in this study are evaluated based on their functional relevance into three groups of (1) genes involved in metabolic changes, (2) genes involved in cell wall changes, and (3) signal transduction which are discussed below (Table 1).

Genes involved in the latent stage
Genes Involved in Metabolic Changes in the Latency Stage
One of the abilities of MTB strain is its quick adaptability with the environment [17]. A considerable number of studies demonstrate that metabolic pathways in MTB changed after entering the macrophage [18]. The presence of MTB in macrophages leads to granuloma formation in human’s lung. The presence and survival of MTB in lung granulomas has been investigated in several studies, and it has been shown that MTB is likely to be present in the environment with conditions such as oxygen depletion, redox stress, increased carbon dioxide, nutrient degradation, and pH reduction [19]. In aerobic conditions of the MTB, the oxidative phosphorylation tricarboxylic acid (TCA) cycle is used to generate energy from the pathway of the electron transfer channel (ETC) for the proton transfer energy. In anaerobic conditions, which reduce oxygen levels, the bacteria will not be able to use conventional pathways to generate energy and use the ETC pathway to transmit electrons [20,21]. The adaptation of MTB to this condition requires the expression of different genes, in order to activate the appropriate metabolic pathways to supply the energy it needs. Modifying the metabolic process requires the alteration of the produced enzymes. The production of enzymes required for changes in the metabolic process necessitates altering the expression of genes involved in the production of these enzymes. In this section, the expression of icl, glcB, gcvB, narGHJI, narK2, and narX genes in latency and their effect on mycobacterial viability are investigated [20,21]. In Figure 1, it has been attempted to illustrate the modification of metabolic pathways and thus the alteration of production of the required enzymes in the metabolic cycles, which are caused by the expression of these genes.
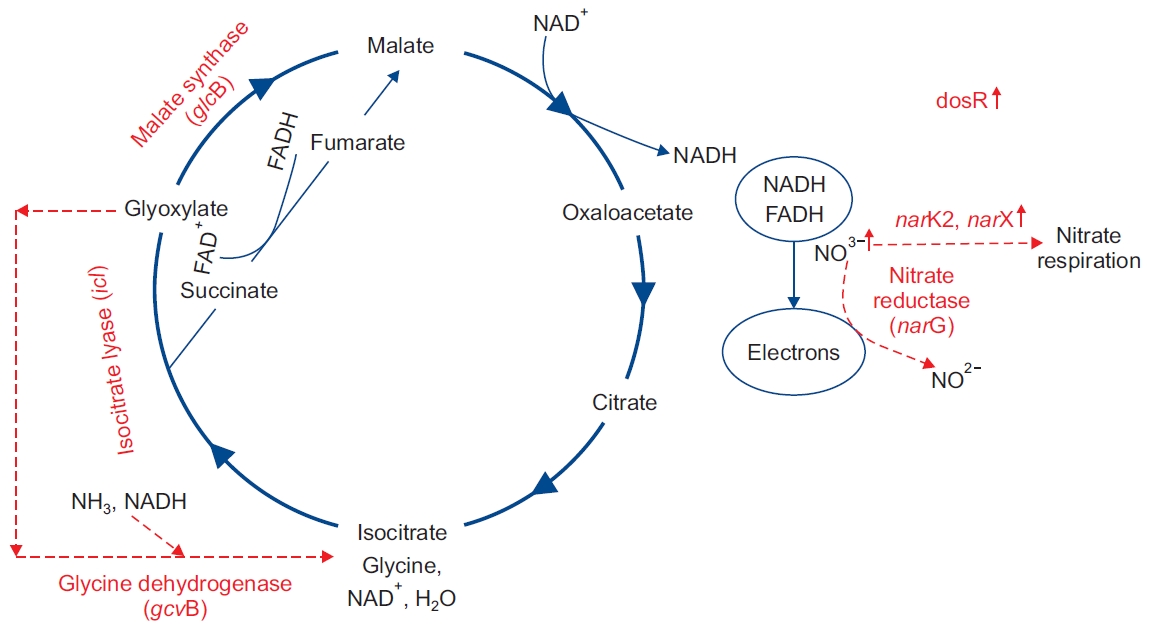
Metabolic changes induced by hypoxia. Enhanced expression of the icl gene caused the conversion of isocitrate to glyoxylate and succinate. The glcB and gcvB genes are involved in the conversion of glyoxylate to malate and glyoxylate to glycine, respectively. The electrons generated in this pathway may lead to nitrate reduction, which is also associated with increased dosR expression. Increased dosR expression also affects the expression of other genes such as narK2 and narX.
1. icl gene
The icl gene MTB encodes the isocitrate lyase (ICL) [22], which is the primary enzyme in the glyoxylate (GLX) cycle [23]. icl gene characteristics of the gene are summarized in Table 1. This enzyme is necessary for the use of fatty acid by bacteria [24] and important for latency in MTB, which is called “persistence factor” [25]. In 1998s, the icl gene was sequenced for the first time by Cole et al. [22] and has been studied extensively since then. This results in the change of isocitrate to GLX and succinate. When the MTB Δicl mutant was examined, it was shown that its activity had not changed in macrophage resting mode, while it had significantly reduced in active mode [25]. The main source of carbon and energy in MTB is fatty acids, which is provided by beta-oxidation. At the time of the hypoxic, ICL production increases significantly (five times). When the carbon source is reduced, the ICL enables the bacteria to use fatty acids as a source of carbon and energy using GLX [26]. Several studies have shown that the expression of the icl gene for MTB transmission is necessary from critical to stable status [18] (Figure 1). The presence of this gene preserves MTB life in latency conditions and therefore contributes to the persistence of infection in the human’s body [27]. Not only is the ICL a key enzyme in the survival of mycobacteria, but it also is not present in humans, so it can be considered a therapeutic target.
2. glcB gene
Many studies have been done on glcB gene since it was the first identified [28]. The glcB gene codes the malate synthase, which leads to bypass of GLX, together with the production of the ICL enzyme by the icl gene [29]. This probably enables the MTB pathway to adapt itself to anaerobic conditions and nutrient deficiencies. These two enzymes also provide the exchange of isocitrate to malate, which helps to keep continuing the TCA cycle [30]. However, studies have shown that there was no significant increase in the production of this enzyme under hypoxic conditions [29] (Figure 2). Since this enzyme is involved in the GLX, and this anaplerotic pathway is not present in mammals, it can be investigated as an option for treatment of tuberculosis.

TreS enzyme functions in the Mycobacterium tuberculosis cell wall structure. This enzyme catalyzes the reversible interconversion of maltose and trehalose. Maltose is converted to maltose-1 phosphate by Mak and trehalose is converted to trehalose monomycolate (TMM) by CmrA, which crosses the cell membrane via MmpL3. Antigen 85 (Ag85s) complex enzymes catalyze the conversion of TMM to trehalose dimycolate (TDM), which plays a significant role in protecting cells under stress.
3. gcvB gene
gcvB gene codes glycine dehydrogenase (GDH), which most likely encodes the P protein of the glycine cleavage system (GCS) [31]. The function of this enzyme, first identified in MTB in 1962, was detected in nonreplicating persistent (NRP) MTBs [32]. As gcvB and gcvH are located in the operon associated with glycine catabolism, the GCS may use glycine as a source of nitrogen [33]. Increasing the expression of this enzyme in hypoxia conditions in non-proliferative MTB is shown [8]. The GDH catalyzes the amination of glyoxalate to glycine and simultaneously catalyzes the oxidation of NADH to NAD [8]. The activity of GDH has been shown to be stable in vitro [32]. Although it showed that the gcvB gene is involved in the expression of the GDH enzyme, other genes are also involved in this process, and further study of the interaction of these genes on this enzyme is needed.
4. narGHJI gene
This gene codes nitrate reductase [34]. Although nitrate reductase enzyme activity encoded by this gene is low in aerobic conditions, it increases in microaerophile [35]. Such a function may be related to the compliance of MTB in a permanent condition [36]. However, it has been shown that hypoxic conditions do not affect narGHJI expression [36]. It is noteworthy that even though nitrate reductase is unaffected, when oxygen is gradually depleted (over a period of more than 46 days), nitrate release reductase increases in the event of an immediate discharge of oxygen [36]. Nitrate is reduced to nitrite by nitrate reductase to produce bacterial energy to enter the NRP step [29]. It has been shown that the activity of nitrate reductase in MTB leads to the continuation of the electron transfer chain under microaerophilic or anaerobic conditions [37]. In addition to the narGHJI gene, the narK2 and narX genes are also involved in nitrate reduction, which are discussed below.
5. narK2 gene
nark2 gene encodes the transporter nitrate, which is expressed in hypoxic conditions [38]. The release of nark2 in hypoxia conditions probably increases nitrite production in MTB [39]. Unlike narGHJI, it has been shown that narK2 is produced in response to the presence of nitrate or nitrite [40]. Another contrast with narGHJI is that, unlike narGHJI, the expression of narK2 increases in hypoxia and decreases in nitric oxide levels [41]. This increase in activity is probably due to the fact that the hypoxia and the presence of NO lead to the inactivation of cytochrome oxidase and the activation of narK2 [42]. It has been shown that ΔnarK2 mutant does not affect the expression of hypoxia [41]. The NarK2 protein is positioned on the surface of the membrane, and can detect the oxygen level of the environment as a sensor, which has been shown to maintain the amount of ATP in hypoxia conditions [40]. The DosR/DevR controller transcribes narK2 in hypoxic situations and reduces NO levels [43].
6. narX gene
narX gene encodes fused nitrate reductase [31]. The studies have shown that narX is homologous with nitrate reductase proteins in other prokaryotes [31]. Although the function of narX has not been completely detected, studies have shown that the expression of this gene in the presence of NO increases over 800 times [44]. NarX is a membrane protein which is likely to contribute to the electron transport chain [45]. It has been suggested that in the absence of oxygen, NarX in the environment can lead to respiratory depression of nitrate as an electron transfer [44]. It has been determined that narX is the first gene to be replicated in latent MTB [45]. Therefore, it is suggested that the expression of this gene be used as a marker for latent MTB diagnosis [44]. However, nark2 is expected for induction of nitrates reductase activity in anaerobic conditions, but narX is not required [44]. Nitrate reduction is one of the activities performed under hypoxia at the latency stage, and the investigation of the genes involved in this activity is important to better understand the latency process.
Genes Involved in Cell Wall Changes in the Latency Stage
One of the characteristics of MTB is the unique cellular wall, due to which the bacterium inside the cell survives. The significant part of this structure are the lipids, and mycolic acid consists of 60% of the cell wall lipids. In the cell wall, there are high molecular weight fatty acids (60–90 carbon), which are common to all mycobacteria species [46]. One of the unique characteristics of MTB cell cover is the presence of 2-alkyl, mycolic acid, and 3-hydroxy fatty acids. The cellular structure of MTB prevents the immune system response to the bacteria. Furthermore, MTB cell wall compounds make it flexible and fluid. The formation of granuloma is one of the reasons for MTB survival, so MTB is stationary in the latent state of granuloma. One of the reasons for the formation of granuloma is the presence of the structure of trehalose-6,6’-dimycolate (TDM), which is also known as the cord-factor [47]. TDM is identified by a receptor called the minacle located on the macrophage, which creates granuloma with NO production [48]. Another effect of this lipid is its inhibitory effect on the migration of leukocytes, which plays a significant function in the survival of the bacteria. In addition to TDM, latent changes occur in the MTB cell wall, which is associated with a change in the expression of the genes related to it [49]. These genes are briefly discussed in the following sections.
1. pcaA
The pcaA gene codes the cyclopropane synthase enzyme [50]. The activity of this enzyme is required for cyclopropanation of mycolic acid [51]. PcaA phosphorylation leads to a decrease in the synthesis of cyclopropane, involving the exchange of mycolic acid profiles [52]. One of the effects of changing the mycolic characterization profile is the stoppage of intracellular bacterial replication and the absence of formation of a phagosome maturation block (PMB) [52]. The ΔpcaA mutant study has revealed that this mutant does not have the ability to produce any serpentine cords and not only does not lead to persistence in mouse models but also results in the death of the mouse [52]. As mentioned before, PcaA is implicated in the formation of PMB and mycolic acid, which leads to the survival of MTB in the host macrophages in latent stage [51]. The results of the studies indicate that pcaA gene activity is required for mycobacterial cell survival and is needed for the escape of macrophage killing.
2. treS
treS gene encodes the trehalose synthase (TreS) enzyme [53]. TreS substrates are trehalose or maltose, but the ability to produce trehalose from maltose is 2.5 times [54]. Trehalose is located in the cytoplasm and glycolipids cell wall of mycobacterium [55]. This compound not only acts as a virulence factor, but also has many other roles [54]. Among different roles of trehalase is the one that it can be used as carbon and energy source as well as its inhibitory effect on bacterial drying and freezing [56]. Although treS removal has no effect on the amount of intracellular trehalose, its expression increases the amount of trehalose in the cell, which indicates the role of this gene in the concentration of intracellular maltose [54]. One of the structures of trehalose is TDM, which is one of the important pathogenicity factors (Figure 2) [54]. As previously stated, TDM plays a pivotal role in the survival of MTB by formation of granuloma and inhibiting the formation of phagosome lysosome fusion [54]. Since trehalose plays both a major role in the cell wall structure and protects mycobacteria under environmental stress conditions, investigating the genes involved in trehalose utilization pathways might be useful for better understanding the latency stage.
Signal Transduction and Latency
Signal transduction is one of the factors affecting MTB survival in a granuloma, which contributes to bacterial survival under conditions such as acidity decrease, nutrient deficiencies, and environmental oxygen depletion [57]. In this review, two-component systems (TCS) and sigma factors are examined in detail.
1. Two-component systems
Eleven TCS have been identified in MTB, which is far lower than other bacteria, including Escherichia coli (more than 30) [58]. TCS in Mycobacterium has several physiological functions, but these functions are not completely understood [59]. One of the functions of TCS is to help maintain bacterial living under environmental degradation conditions (such as macrophage environment or antimicrobial agents), and TCS is an extremely effective factor in bacterial pathogenesis [60]. The TCS includes sensor histidine kinase (HK), which, using phosphorylation cognate response regulator (RR), regulates the expression of the gene and creates an appropriate response to the sensor [59]. A significant characteristic of TCS, its presence in all prokaryotes and its nonbeing in high-level eukaryotes, has made it a valuable treatment option [61]. It has been shown that the amount of pathogenicity in mutant lacking TCS considerably decreases [58]. TcrX and TcrY of open reading frames play an important role in coding RR and HK, respectively [62].
1) dosR
The dosR regolone consists of approximately 50 genes (Table 2) [63,64], which are essential for survival in the latent period [65]. DosR is a transcritical factor, which plays a significant role in adapting to the initial conditions of hypoxia, reducing NO and carbon monoxide levels [66]. It has been revealed that DosR leads to the regulation of the TAG (TAG production gene tgs1) gene, Tgs (TAG), which also stops the production of TAG by removing DosR, and the increase of TAG production by provoking DosR [67]. The DosR regolon promotes the production of antigens, which are identified by T-cells at latency, and are essential for controlling infection. It is likely that T-cell response is due to the induction of interleukin (IL)-10, IL-17, and interferon-γ production in the latent condition [68].
Genes regulated by dosR under hypoxic conditions
In this two-compound system, DosS and DosT act as HK, which activate DosR [69]. Transcription of the dosR, dosS genes occur together, possibly due to the genetic association of them [70]. The dosR and dosS genes are considered preserved genes, while the dosT gene is preserved less [71]. Studies have shown that DosR activity is necessary for the adaptation of MTB in hypoxia conditions [69]. It is also observed in studies that, despite the structural similarities in DosT and DosS, these proteins are functionally different, so DosT acting as a direct oxygen sensor and DosS as a redox sensor [66]. Since the expression of dosR regolon increases in the latency stage, and T cells show specific responses to some of the antigens which are specific to the regolon, it is considered a candidate for the production of a vaccine and drug.
2) phoP gene
The phoP gene codes the transcriptional regulator of the TCS PhoPR. PhoP has been distinguished as having several functions that appear to be essential during latency to keep the bacteria alive [72]. phoP leads to the regulation of the expression of the genes of pks2 (induction of sulfolipids production), pks3 (induction of polyacyltrehalose/diacyltrehalose production) and ald (induction of L-alanine dehydrogenase production) [73]. This phoP also provides the metabolism of cell wall fats, the regulation of the intestinal acidity of the cell, the adaptation to the conditions of thermic stress, and the response to the early stages of hypoxia in MTB [74]. Another significant point is that of the expression of the icl gene is negatively regulated by phoP. It has been designated that although ΔphoP mutant does not have the capability to replicate, it can survive in the macrophage [72]. Although the phoP gene is required for mycobacterial intracellular growth, the need for it in the latency is still controversial.
3) mprA
The mprA gene encodes the two-component regulator [31]. MprA is part of the MtrA-MtrB system, which was the first TCS system to be distinguished in the 1990s [75]. This system is involved in controlling MTB replication in macrophages and regulating significant processes of MTB physiology [57,76]. Studies have shown that the expression of the mprA gene is effective in maintaining latency infection in MTB so that the ΔmprA mutant does not have the ability to keep the infection in a latent status [77]. The role of MprA in regulating the expression of the DosR regolum and Rv1813, which is involved in regulating the expression of type II NADH dehydrogenase, is shown in some unfavorable environmental conditions [78]. Identification of regulatory functions of MprA leads to a better understanding of the mechanisms involved in latency stage in the host and will ultimately lead to a better understanding of how to establish TB control in the latency stage.
4) kdpDE
The two-component kdpDE system is expressed by the Rv1027c and Rv1028c genes [79]. It has been reported that kdpDE TCS is present in many species of bacteria (more than 1,000 species), which are extensively protected [80]. The expression of kdpDE increases in incompatible conditions such as reduced nutrients, temperature stress, acidity, and hypoxia. In vivo, kdpD expression increases in nutrient depravity conditions, and expression of kdpE also increases during growth in macrophages [81]. Both kdpD and kdpE genes are located on an operon, and they are expressed in a particular variant. The position of phosphorylation in KdpD and KdpE is His642 and Asp52, respectively, where the phosphorylation of KdpD and the transfer of the phosphoryl group to KdpE leads to the response to fluctuations in the acidity of the environment [79]. The results of our studies indicate that kdpDE leads to a decrease in mycobacterial growth within the macrophage, which may be due to a change in the mycobacterial cell wall. This function can be important in preserving mycobacteria in the latency phase.
5) tcrY
The tcrY gene encodes the two-component regulator [31]. tcrY and tcrX are implicated in the formation of TCS TcrX/Y in MTB [82]. This system is protected in all species of Mycobacterium except Mycobacterium leprae [62]. It has been confirmed that in mouse models where tcrX/Y is eliminated, the severity of the disease increases, and the mouse model dies earlier [83]. TcrY contains three domains, including the C-terminal located in the outer the cell, a membrane domain, and the N-terminal part positioned in the space of cytosol [82]. TcrX is also generally located in the cytosol section [84]. TcrY is phosphorylated in the presence of Mg2+ or Ca2+ ions, which passes this phosphorus to TcrX [84]. It is shown that tcrY and tcrX are apparently translated in one pathway [84]. Although the results of the present study indicate that tcrY is expressed under conditions of iron restriction and post-infection, little is known about the conditions affecting the expression of this two-component regulator; therefore, further studies are needed to investigate the effect of this gene on latency.
6) trcSR
The trcSR TCS is coded by trcS and trcR, respectively [85]. In this TCS, TrcS with phosphorylation in the presence of Mg2+ or Ca2+ ions leads to the transfer of phosphorus to TrcR [86]. Despite studies of the function of this system, the only evidence that indicates the expression of TCS trcSR in latency is that the system is expressed at an early stage of infection in macrophages and in conditions of reduced oxygen in the environment [84]. However, its precise function is still not completely defined [86].
2. Sigma factors
Sigma factors are present in all bacteria except in Mycoplasma. They also regulate expression, therefore responding to environmental stress requires the formation of a network of factors so that the bacteria can survive. In addition, the reprogramming of RNA polymerase is required to translate the factor σ [87]. In general, sigma factors are divided into two main groups, which include σ54 and σ70 [88]. In MTB, there is 1 (sigma) of the ‘housekeeping’ factor and 12 σ ancillary factors. Housekeeping σ factor is expressed in response to natural conditions, while σ ancillary factors are expressed in response to environmental stresses [89]. All of the σ factors available in the MTB are σ70, and the presence of this type of σ factor has been confirmed in all bacterial species [88,90].
1) sigF
SigF expression occurs at the stationary stage as well as in the presence of environmental stress such as cold stress, nutrient depletion, acidity change, and the presence of drugs, particularly metronidazole [89]. SigF expression is regulated by Usfx [91]. In addition to being directly involved in the expression of 14 other genes, SigF is also engaged in the expression of other sigma factors. For example, it influences on viral virulence by affecting the expression of SigC [92]. Identification of genes involved in preserving bacterial life at the late stage of growth important to eliminate MTB at this stage because it can help identify the therapeutic process.
2) sigH
Sigma H is expressed in response to high-temperature stress, oxidative thiol, and macrophage entry [22,93-95]. sigH gene, like most of the genes examined in this study, was identified by the MTB genes sequence in the 1980s [22]. SigH is implicated in the translation of 31 structural genes, leading to the induction of sigB and sigE [96]. It is acknowledged that sigH is expressed when MTB enters the host cell, so the expression of this factor may be effective in provoking latency [97]. Thus, many studies have determined that Δ-σH mutant in the murine model will not be able to induce normal granuloma [95]. sigH gene studies indicate that it plays an important role in the regulation of heat and oxidative stress response and is probably important in the pathogenesis of MTB.
3) sigE
SigE is involved in the expression of genes that lead to pathogenicity which has been studied in many studies [95]. SigE is expressed in response to oxidative stress, sodium dodecyl sulfate detergent, and high temperature, so the viability of sigE mutants would be much lower under these conditions, and its growth decreases in the granuloma and mouse model [95]. Studies have also confirmed that the induced immune response to the sigE mutant would be more effective in the BALB/c mouse model, making this sigma a candidate for the vaccine [95,98]. Therefore, as a therapeutic option, further understanding of this gene’s function and its implications seems to be a necessity.
4) whiB
WhiB3, a cytoplasmic redox sensor, is required to induce MTB resistance under acidic stress conditions [99]. WhiB3 regulates the expression of genes engaged in responding to decreased acidity, hypoxia, NO reduction, redox metabolism, and lipid anabolism [100]. It has been revealed that the MTBΔwhiB3 mutant greatly reduces its virulence severity in the mouse model. WhiB expression is induced during the early stage of rat lung infection and resting macrophages, but is suppressed in activated macrophages [99]. Although the function of whiB has been demonstrated in situations of changes in oxygen concentration, its precise role in MTB adaptation to these changes has not been elucidated, so further investigations in this area are essential.
Conclusion
“Latency”, “persistence”, and “dormancy” are common terms which are used interchangeably in related studies. When a person is infected by MTB, the bacterium is capable of persisting during a prolonged period in a process named LTBI. Conventionally, LTBI has been known to engage in the bacilli lasting in a non-duplicating state in old lesions but still maintaining their capability to cause reactivation and active TB upon occurrence of an immune response disturbance.
In this study, we attempted to induce the distinguished latency genes in MTB. Understanding the latency requires the identification of genes that can sustain MTB to survive in hypoxia, nutrient degradation, environmental stress, and acidity variations. Distinguishing these genes allows researchers to be able to study treatment options and new vaccines more precisely. Although many studies have been done to identify these genes, there are still many uncertainties about this issue. Therefore, further studies in this field are required in the laboratory environment and animal models to shed more light on the latency process.
Notes
Authors’ Contributions
Conceptualization: Hamidieh F, Farnia P, Nowroozi J, Farnia P, Velayati AA. Validation: Farnia P, Velayati AA. Investigation: Farnia P, Hamidieh F. Writing - original draft preparation: Hamidieh F. Writing - review and editing: Farnia P, Nowroozi J, Farnia P. Approval of final manuscript: all authors.
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Funding
The project approved by scientific committee and funded by National Research Institute of Tuberculosis and Lung Disease (NRITLD), IR.SBMU.NRITLD.REC.1397.566.
Acknowledgements
This article is sponsored by Dr. Masih Daneshvari Hospital, Mycobacteriology Research Center (MRC), National Research Institute of Tuberculosis and Lung Disease (NRITLD) and they are thanked.