Role of AMP-Activated Protein Kinase (AMPK) in Smoking-Induced Lung Inflammation and Emphysema
Article information
Abstract
Background
AMP-activated protein kinase (AMPK) not only functions as an intracellular energy sensor and regulator, but is also a general sensor of oxidative stress. Furthermore, there is recent evidence that it participates in limiting acute inflammatory reactions, apoptosis and cellular senescence. Thus, it may oppose the development of chronic obstructive pulmonary disease.
Methods
To investigate the role of AMPK in cigarette smoke-induced lung inflammation and emphysema we first compared cigarette smoking and polyinosinic-polycytidylic acid [poly(I:C)]-induced lung inflammation and emphysema in AMPKα1-deficient (AMPKα1-HT) mice and wild-type mice of the same genetic background. We then investigated the role of AMPK in the induction of interleukin-8 (IL-8) by cigarette smoke extract (CSE) in A549 cells.
Results
Cigarette smoking and poly(I:C)-induced lung inflammation and emphysema were elevated in AMPKα1-HT compared to wild-type mice. CSE increased AMPK activation in a CSE concentration- and time-dependent manner. 5-Aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR), an AMPK activator, decreased CSE-induced IL-8 production while Compound C, an AMPK inhibitor, increased it, as did pretreatment with an AMPKα1-specific small interfering RNA.
Conclusion
AMPKα1-deficient mice have increased susceptibility to lung inflammation and emphysema when exposed to cigarette smoke, and AMPK appears to reduce lung inflammation and emphysema by lowering IL-8 production.
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by persistent airflow limitation that is associated with an enhanced chronic inflammatory response in the airways and lungs to noxious particles or gases1. Cigarette smoking is the most important risk factor for the development of COPD. Chronic smoke exposure causes airway and lung parenchymal inflammation which leads to small airway disease (obstructive bronchiolitis) and emphysema. The biologic nature of COPD inflammation is not well understood, and pharmacological agents that inhibit inflammation in COPD are urgently needed. The so-called traditional hypothesis states that COPD is initiated and maintained by the activation of inflammation by long-term cigarette smoking, leading to a protease/anti-protease imbalance2. Though this hypothesis has compelling experimental and observational clinical support, it does not address several observations particularly related to the progressive nature of COPD: the several decades of smoking required to develop the disease3, the persistence of inflammation despite smoking cessation4, and the vexing observation that corticosteroids have little impact in COPD5. Additional hypotheses for the pathogenic mechanism of COPD include oxidative stress, auto-immunity, apoptosis and senescence6,7,8,9,10,11. More research on the cellular and molecular mechanisms that regulate the development and progression of COPD is required to identify novel therapeutic targets.
AMP-activated protein kinase (AMPK) is a well-known energy sensor that is activated by increased intracellular AMP levels12. AMPK activation is initiated by alterations in cellular metabolic state that result from inhibition of ATP generation, such as hypoxia, glucose deprivation, heat shock, or reduction in mitochondrial oxidative phosphorylation, or from increased ATP consumption, such as occurs during exercise or when cellular metabolism increases. AMPK not only functions as an intracellular energy sensor and regulator, but is also a general stress sensor that is important in maintaining intracellular homoeostasis during oxidative stress and other potentially damaging situations13,14. Furthermore, there is also evidence that it reduces acute inflammatory reactions, apoptosis, and cellular senescence15,16,17,18,19,20,21,22,23,24,25,26. Based on these findings, we reasoned that AMPK might be involved in limiting COPD pathogenesis. In the present study, we sought to investigate its possible role in modulating cigarette smoking-induced lung inflammation and emphysema. To examine these issues, we first compared cigarette smoke- and polyinosinic-polycytidylic acid [poly(I:C)]-induced lung inflammation and emphysema in AMPKα1-deficient (AMPKα1-HT) mice and wild type mice of the same genetic background. We subsequently investigated the role of AMPK in the induction of interleukin (IL)-8 by cigarette smoke extract (CSE) in A549 cells.
Materials and Methods
1. Reagents and antibodies
We purchased poly(I:C) from InvivoGen (San Diego, CA, USA). 5-Aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR), an activator of AMPK and Compound C, an inhibitor of AMPK, were purchased from Merck Millipore (Darmstadt, Germany). For immunoblot analysis, antibodies against β-actin, AMPK-α, phospho-AMPK-α, and microtubule-associated protein 1 light chain 3-I/II were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-IL-8 and tumor necrosis factor α(TNF-α), antibody were from Santa Cruz Biotechnology (Dallas, TX, USA).
2. Mice
Heterozygous AMPKα1-deficient (AMPKα1-HT) C57BL/6 mice were generously provided by Professor Benoit Violet (INSERM U567, Paris, France) and bred in specific pathogen-free conditions in the facility of the Asan Institute for Life Science. Animal experiments were approved by the Institutional Animal Care and Use Committee of Asan Medical Center, Seoul, Korea.
3. Genotyping of mice
Routine genotyping was carried out by multiplex polymerase chain reaction with primers synthesized by Bioneer (Daejeon, Korea). Reaction conditions were as follows: 5 minutes at 94℃, 35 cycles of 94℃ for 30 seconds, 60℃ for 30 seconds and 72℃ for 1 minute, and 5 minutes at 72℃. Primers were as follows: for AMPKα1 knockout, forward (F) 5'-GGGCTGCAGBAATTCGATAT CAAGC-3' and reverse (R) 5'-CCTTCCTGAAATBACTTCTG GTGC-3', and for AMPK wild type, forward (F) 5'-AGCCGACT TTGGTAAGGATG-3', and reverse (R) 5'-CCCACTTTCCATTT TCTCCA-3', yielding amplification products of 350 bp and 450 bp, respectively (Figure 1A).

Generation of heterozygous AMPKα1-deficient (AMPKα1-HT) C57BL/6 mice. (A) Genotyping of mice by multiplex polymerase chain reaction; this yielded amplification products of 350 bp (knockout) and 450 bp (wild-type). (B) Western blot analysis of AMPKα expression in lung tissue. AMPK: AMP-activated protein kinase; WT: wild-type littermate; HT: heterozygous AMPKα1-deficient mouse; M: marker. Data represent mean±SEM (n=3). Each data point is based on three independent Western blots. *p<0.05, vs. WT mice.
4. Exposure of mice to cigarette smoke, and administration of poly(I:C)
Mice were exposed to the mainstream smoke of 20 filtered commercial cigarettes per day, each containing 8.0 mg tar and 0.8 mg nicotine (This Wild; KT&G, Daejeon, Korea). The mice were exposed to cigarette smoke 5 days per week for 2 months. Ten to twelve mice were settled in an inhalation box (50×40×30 cm) connected to a pump and were exposed for 10 minutes to mainstream cigarette smoke generated simultaneously from 10 cigarettes. The mice remained in the box for an additional 10 minutes after the cigarettes had burned out. The box was then ventilated to remove the cigarette smoke, and the mice breathed normal room air for 5 minutes. A second exposure was performed in the same manner. Thereafter the mice were returned to their cages. Control animals inhaled clean room air only in the cages. After 2 weeks of cigarette smoke exposure, 50 µL poly(I:C) or their vehicle controls were administered twice per week (every Monday and Thursday) for 2 weeks (days 15, 18, 22, and 25, respectively) via nasal aspiration. Cigarette smoke exposure was continued during this period.
5. Preparation of bronchoalveolar lavage fluid cells and lung tissues
At the end of each experiment, mice were anesthetized with isoflurane and a middle thoracotomy was performed. The trachea was cannulated and perfused with three 0.7-mL aliquots of cold saline. The heart and lungs were then removed en bloc . The left main bronchus was ligated, and the right lung was inflated by intratracheal infusion of 0.3% low-melting agar at 25 cm H2O. This allowed for homogenous expansion of the lung parenchyma, as described previously27. The inflated right lung was fixed with 4% paraformaldehyde and embedded in paraffin, and the left lung was stored at -80℃ for subsequent analysis The samples of bronchoalveolar lavage (BAL) fluid were centrifuged at 350 ×g for 5 minutes at 4℃, and the supernatants were stored at -80℃ for later analysis. The cell pellets were re-suspended in phosphate buffered saline to count cells.
6. AMPK activity in lung tissue
AMPK activation was assessed as the ratio of phospho-AMPK to total AMPK after correction for local background intensity and normalization to β-actin. Lung tissue was homogenized in lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1mM β-glycerophosphate, 50 mM NaF, 1 µM Na3VO4, 1 µg/mL of leupeptin, 10 µg/mL aprotinin, and 1 mM AEBSF). Lung tissue proteins were analyzed by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting with mouse monoclonal anti-total AMPK-α antibodies (Cell Signaling Technology) and rabbit monoclonal anti-phospho-AMPK-α at thr172 (Cell Signaling Technology).
7. Lung morphometry
Lung sections of 4-mm thickness were used for histological analysis. Mean linear intercepts (MLI) were determined on paraffin-embedded lungs by staining with hematoxylin and eosin, as described previously28. All histological examinations were made by two investigators in a blinded manner.
8. Cell culture
Alveolar type II-like epithelial cell line A549 (#CCL-185; American Type Culture Collection, Manassas, VA, USA) was maintained in Dulbecco's modified Eagle's medium containing 10% fetal calf serum. A549 cells were cultured for 24 hours in 100-mm tissue culture plates in RPMI 1640 (American Type Culture Collection) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA), 100 U/mL penicillin, and 100 µg/mL streptomycin (Life Technologies Inc., Frederick, MD, USA). The cells were incubated at 37℃ in a humidified atmosphere containing 5% CO2 and 95% air and pretreated with AICAR (1 mM) or Compound C (10 µM) in serum-free RPMI for 5 hours before exposure to 3% CSE for 24 hours.
9. CSE preparation
10% CSE was prepared by bubbling smoke from one cigarette (Eighty-Eight Light containing 8.5 mg tar and 0.9 mg nicotine/cigarette) into 10 mL of serum-free culture medium at a rate of one cigarette per minute as described previously29. The CSE preparation was standardized by monitoring absorbance at 320 nm (optical density of 0.74±0.05)30. CSE solution filtered through an aseptic 0.22-µm filter was taken as 100%.
10. Transfection of AMPKα-1 siRNA
To knockdown AMPKα1 expression, human AMPKα1-specific small interfering RNA (siRNA) duplexes were obtained from Bioneer. A549 cells were transfected with human AMPK a1 siRNA duplexes (10 nM) using Lipofectamine RNAiMAX (Invitrogen) and experiments were performed 48 hours later. A non-silencing siRNA, which did not influence the expression of AMPK, was used as a negative control. After transfection, cells were treated with 3% CSE for 24 hours in serum-free medium.
11. Western blot analysis
Aliquots of cell lysates, tissue lysates, or membrane protein extracts were separated on 18% SDS-PAGE and blotted onto PVDF membranes (Amersham, Buckinghamshire, UK). The membranes were blocked with 5% skim milk in Tris-buffered saline (TBS) with 0.1% Tween 20 (TBST). They were then probed with specific primary antibodies to AMPK, phospho-AMPK, IL-8, and β-actin at 4℃ overnight. Horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG (Bethyl Laboratories, Montgomery, TX, USA) was used to detect primary antibodies. Blots were visualized with ECL reagent (Amersham). Equal loading of the gel was checked by measuring protein as well as by reprobing membranes for β-actin. Image J software (version 1.41; National Institutes of Health, Bethesda, MD, USA) was used for quantitative densitometric analysis.
12. Determination of IL-8 in culture medium
The concentrations of IL-8 in culture medium were measured with a human IL-8 enzyme-linked immunosorbent assay (ELISA) kit (R&D systems Inc., Minneapolis, MN, USA). Cell culture media were centrifuged at 14,000 ×g for 10 minutes at 4℃, and supernatants stored at -80℃ prior to analysis.
13. Statistical analysis
Data are presented as means±SEM. For comparison between groups, the Mann-Whitney U test and Kruskal-Wallis tests were used. Statistical evaluations were undertaken with SPSS statistical package version 15.0 (SPSS Inc., Chicago, IL, USA). Statistical significance was defined at p<0.05.
Results
1. Cigarette smoking and poly(I:C) induce more extensive lung inflammation and emphysema in AMPKα1-deficient (AMPKα1-HT) mice
To elucidate the role of AMPKα1 in cigarette smoke-induced lung inflammation and emphysema, AMPKα1-deficient mice were exposed to cigarette smoke or challenged with poly(I:C). The AMPKα1-deficient (AMPKα1-HT) mice produced approximately 50% of the AMPKα of wild type mice (Figure 1B). Cigarette smoke exposure and poly(I:C) treatment induced significant increases in total BAL fluid cells, and neutrophil and macrophage counts, as well as in MLI, in both wild type and AMPKα1-HT mice. However total BAL fluid cells, lymphocyte and macrophage counts, and MLI were significantly higher in the AMPKα1-HT mice than the wild type mice after exposure to cigarette smoke or poly(I:C) (Figure 2A-C).

Increased cigarette smoking- and poly(I:C)-induced lung inflammation and emphysema in AMPKα1-deficient (AMPKα1-HT) mice. (A) Histological assessment at 2 months of lung sections stained with hematoxylin and eosin (×100). (B) Total bronchoalveolar lavage cell counts and differential cell counts. (C) Morphometric analysis of mean linear intercepts. AMPK: AMP-activated protein kinase; WC: wild-type no smoke (n=4); HC: AMPKα1-HT no smoke (n=5); WS: wild-type exposed to smoke (n=4); HS: AMPKα1-HT exposed to smoke (n=5). Data represent mean±SEM. * and † denotes significant differences (p<0.05) between the WC and WS group, and between the WS and HS group, respectively.
2. CSE increases IL-8 production in A549 cells
Analysis of cell lysates showed that exposure of A549 cells to various concentrations (0%, 1.5%, 3%, and 4.5%) of CSE for 24 hours increased IL-8 protein in a concentration-dependent manner (Figure 3A). CSE exposure also increased the release of IL-8 from A549 cells (Figure 3B). We chose 3% CSE as the standard CSE concentration in subsequent experiments. Exposure of A549 cells to 3% CSE increased IL-8 protein levels and release of IL-8 in a time-dependent manner (Figure 3C, D).
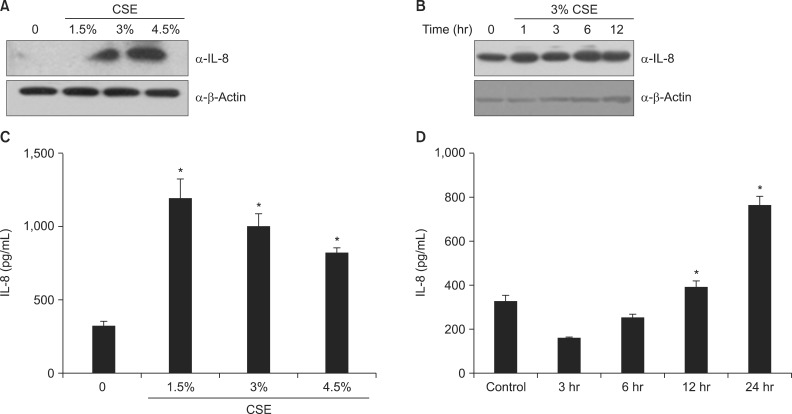
CSE increases the production of IL-8 in A549 cells. (A, C) Cells were exposed to 0%-4.5% CSE for 24 hours. (B, D) Cells were incubated with medium alone (time 0) or exposed to 3% CSE for the times indicated. Protein levels in cell lysates (A, B) were analyzed by Western blot, and protein levels in culture media (C, D) were analyzed by enzyme-linked immunosorbent assay. CSE: cigarette smoke extract; IL-8: interleukin 8. Data in each group are means±SEM of three independent experiments. *p<0.05, vs. 0% CSE (A, C) or time 0 (B, D).
3. CSE increases the phosphorylation of AMPK
Exposure of A549 cells to CSE increased the phosphorylation of AMPK within 60 minutes (Figure 4A). The adenosine analog AICAR, an AMPK activator, also increased the phosphorylation of AMPK, and Compound C, an AMPK inhibitor, inhibited it (Figure 4B, C). Pretreatment with a siRNA specific for AMPK reduced AMPK protein level (Figure 4D).
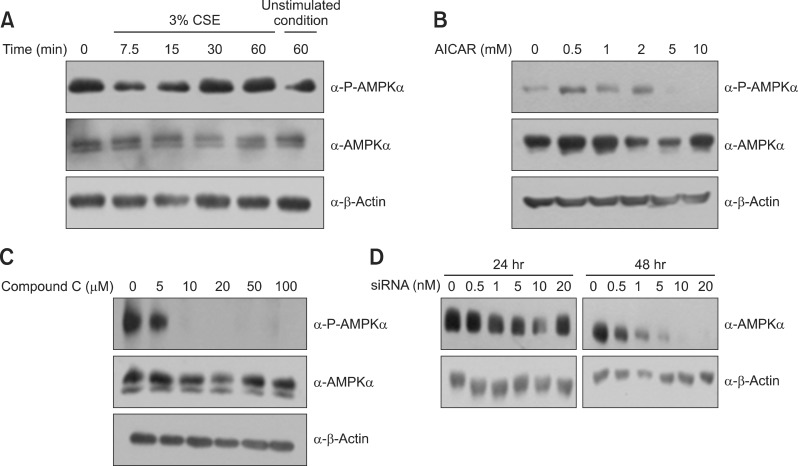
CSE activates AMPK in A549 cells. (A) Cells were incubated with medium alone or exposed to 3% CSE for the times indicated. (B) Cells were exposed to 0-10 mM AICAR (an AMPK activator) for 5 hours. (C) Cells were exposed to 0-100 µM Compound C (an AMPK inhibitor) for 5 hours. (D) Cells were transfected with various concentrations of AMPK siRNA for 48 hours. CSE: cigarette smoke extract; AMPK: AMP-activated protein kinase; AICAR: 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside.
4. Activation of AMPK opposes the release of IL-8 by CSE
To determine whether AMPK is involved in the CSE-induced IL-8 production, A549 cells were pretreated with AICAR (1 mM) and Compound C (10 µM) for 5 hours, and then exposed to 3% CSE for 30 minutes. Cell lysates were subjected to immunoblot analysis and supernatant IL-8 was measured by ELISA. As shown in Figure 5A and C, AICAR decreased the CSE-induced IL-8 production and Compound C increased it. In addition, pretreatment with AMPK siRNA (10 nM) increased the CSE-induced IL-8 production, whereas sham pretreatment or negative (control) siRNA failed to have an effect (Figure 5B, D).
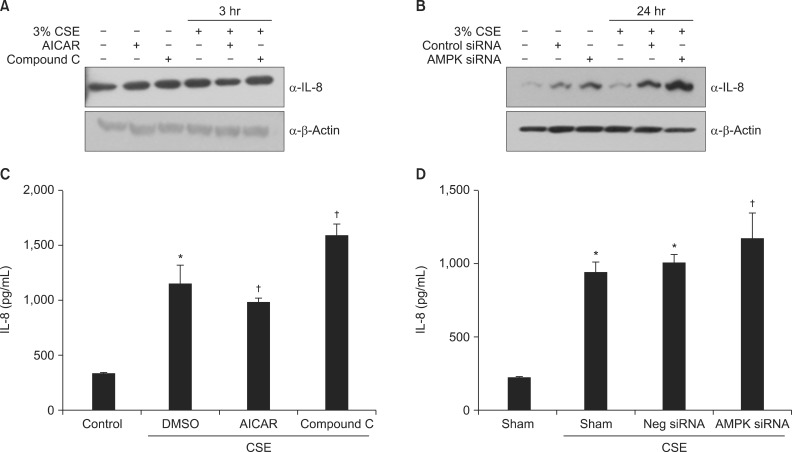
AMPK plays a vital role in the induction of IL-8 by CSE in A549 cells. (A, C) Cells were incubated with medium alone or exposed to 3% CSE for 3 hours after pretreatment with AICAR (1 mM) and Compound C (5 µM). (B, D) Cells were incubated with medium alone or exposed to 3% CSE for 24 hours after pretreatment with 10 nM AMPK siRNA or negative siRNA. Protein levels in cell lysates (A, B) were analyzed by Western blot, and protein levels in culture media (C, D) by enzyme-linked immunosorbent assay. The immunoblot results are representative of three independent experiments. AMPK: AMP-activated protein kinase; CSE: cigarette smoke extract; AICAR: 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside; IL-8: interleukin 8; DMSO: dimethyl sulfoxide. Data in each group are means±SEM (n=3). *p<0.05, vs. medium only. †p<0.05 vs CSE without drug or siRNA treatment.
5. Inhibition of AMPK activation with Compound C increases IL-1 β-induced IL-8 secretion
IL-1β is an important pro-inflammatory cytokine participating in the pathogenesis of COPD. We tested whether AMPK activity also affected IL-1β-induced IL-8 production. IL-1β increased IL-8 secretion in A549 cells. AICAR pretreatment had no effect on secretion, whereas Compound C increased it (Figure 6A, B).

Inhibition of AMPK activation by Compound C increases IL-1β-induced IL-8 secretion in A549 cells. Cells were incubated with medium alone or exposed to 5 ng/mL IL-1β for 24 hours after pretreatment with AICAR (1 mM) or Compound C (10 µM) for 5 hours. (A) Protein levels in cell lysates were analyzed by Western blot. (B) Protein levels in culture media were analyzed by enzyme-linked immunosorbent assay. The immunoblot results are representative of three independent experiments. AMPK: AMP-activated protein kinase; IL-1β, interleukin-1β; AICAR: 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside. Data in each group are means±SEM (n=3). *p<0.05, vs. medium only. †p<0.05, vs. CSE without AICAR or Compound C.
Discussion
To our knowledge, this is the first report that AMPK has an important role in reducing CSE-induced lung inflammation and emphysema. We observed increased cigarette smoke-induced lung inflammation and emphysema in AMPKα1-deficient mice, and in in vitro experiments we showed that inactivation of AMPK by Compound C or AMPK siRNA increased CSE- and IL-1β-induced IL-8 production in A549 cells.
A key mechanism in the pathogenesis of COPD is thought to be an abnormal inflammatory response in the lungs to the inhalation of toxic particles and gases derived from tobacco smoke31. The inflammatory response in COPD involves both innate and adaptive immune responses. Multiple inflammatory mediators are increased in COPD and are derived from inflammatory cells and structural cells of the airways and lungs32. Epithelial cells are provoked by cigarette smoke to produce inflammatory mediators, including TNF-α, IL-1β, IL-6, IL-8 (CXCL8) and granulocyte-macrophage colony-stimulating factor33,34. We also showed that exposure of A549 cells to CSE increased the production of IL-8. A549 cells (adenocarcinomic human alveolar epithelial cells) are widely used as an in vitro model of type II pulmonary epithelial cells. A549 cells release neutrophil and monocytic chemotactic factors such as, IL-8, granulocyte colony-stimulating factor, leukotriene B4 and monocyte chemoattractant protein (MCP-1) in response to CSE35. IL-8 (CXCL8) is a potent neutrophil chemoattractant released by a variety of lung cells, including epithelial cells, macrophages, T cells and neutrophils themselves. It is a key chemokine in CSE-induced lung inflammation; its concentration is increased in the induced sputum of COPD patients and rises further during exacerbations36,37. Although we demonstrated that inactivation of AMPK by Compound C or AMPK siRNA increased CSE and IL-1β-induced IL-8 production in A549 cells, the number of neutrophils in BAL fluid after exposure to cigarette smoke or poly(I:C) did not differ between AMPKα1-deficient and wild-type mice. In a recent study it was shown that activation of AMPK with metformin or berberine followed by stimulation with W-peptide, a mouse homolog of human IL-8, resulted in enhanced mouse neutrophil chemotaxis whereas stimulation with W-peptide led to a decrease in human neutrophil mobility. Thus our results may be explained by decreased neutrophil chemotaxis of AMPKα1-deficient mice38.
Previous evidence suggests that AMPK affects inflammation both positively and negatively. In contrast to our findings, Tang et al.15 reported that preventing AMPK activation with Compound C or AMPK siRNA reduced CSE-induced IL-8 production in human bronchial epithelial cells and lung inflammation in mice exposed to cigarette smoke for 1 month. However, several groups have reported that activation of AMPK by pharmacological activators inhibits inflammatory responses in human liver cells, bronchial epithelial cells, and murine macrophages and neutrophils16,17,18,19. We also previously found that AICAR, an AMPK activator, attenuated poly(I:C)- and ovalbumin-induced airway inflammation and activation of nuclear factor κB (NF-κB), and the production of TNF-α in a murine model of asthma20. Furthermore, AMPKα1-deficient mice showed enhanced airway inflammation and fibrosis in response to challenges by both allergens and bleomycin, while metformin prevented airway inflammation and airway remodeling in a murine model of chronic asthma, at least in part by activating AMPK21. Thus, AMPK may promote or suppress inflammation induced by inflammatory stimuli depending on the cell type. AMPK activation may inhibit inflammation by multiple pathways, notably by inhibiting the NF-κB pathway. For example, AMPK activation in lipopolysaccharide-treated neutrophils was associated with decreased degradation of IκB and diminished nuclear translocation of NF-κB, as well as reduced release of TNF-α and IL-622.
There is evidence that AMPK is an oxidative stress sensor and redox regulator in addition to its traditional role as an energy sensor and regulator. An imbalance between oxidants and antioxidants occurring in COPD results in oxidative stress, which underlies many of the pathogenic mechanisms in COPD. Several studies indicate that activation of AMPK inhibits oxidative stress39,40. AMPK can be activated by increased intracellular reactive oxygen species (ROS) and reactive nitrogen species. Activated AMPK appears to be essential for maintaining intracellular redox status by inhibiting oxidant production by NADPH oxidases or by increasing the expression of antioxidant enzymes. Oxidative stress-induced activation of AMPK is believed to be important for the beneficial effects of many drugs. AMPK regulates the ROS/redox balance, autophagy, cell proliferation, cell apoptosis, cell polarity, mitochondrial function and genotoxic responses, either directly or indirectly, via numerous downstream pathways. Further investigations are required to clarify the exact molecular targets of AMPK under physiological and pathological conditions, and to develop potential strategies to modulate AMPK activation in COPD.
In conclusion, we demonstrate a novel role of AMPK in reducing lung inflammation and emphysema. AMPKα1-deficient mice have increased susceptibility to lung inflammation and emphysema in response to cigarette smoke. We also present evidence that AMPK reduces lung inflammation and emphysema by reducing IL-8 production. Further studies are required to expand our understanding of the role of AMPK in the pathogenesis of COPD, particularly with respect to limitation of CSE-induced lung inflammation.
Acknowledgements
The Korean Academy of Tuberculosis and Respiratory Diseases provided financial support for this research.
Notes
No potential conflict of interest relevant to this article was reported.