EphA2 Receptor Signaling Mediates Inflammatory Responses in Lipopolysaccharide-Induced Lung Injury
Article information
Abstract
Background
Eph receptors and ephrin ligands have several functions including angiogenesis, cell migration, axon guidance, fluid homeostasis, oncogenesis, inflammation and injury repair. The EphA2 receptor potentially mediates the regulation of vascular permeability and inflammation in response to lung injury.
Methods
Mice were divided into 3 experimental groups to study the role of EphA2 signaling in the lipopolysaccharide (LPS)-induced lung injury model i.e., IgG+phosphate-buffered saline (PBS) group (IgG instillation before PBS exposure), IgG+LPS group (IgG instillation before LPS exposure) and EphA2 monoclonal antibody (mAb)+LPS group (EphA2 mAb pretreatment before LPS exposure).
Results
EphA2 and ephrinA1 were upregulated in LPS-induced lung injury. The lung injury score of the EphA2 mAb+LPS group was lower than that of the IgG+LPS group (4.30±2.93 vs. 11.45±1.20, respectively; p=0.004). Cell counts (EphA2 mAb+LPS: 11.33×104±8.84×104 vs. IgG+LPS: 208.0×104±122.6×104; p=0.018) and total protein concentrations (EphA2 mAb+LPS: 0.52±0.41 mg/mL vs. IgG+LPS: 1.38±1.08 mg/mL; p=0.192) were decreased in EphA2 mAb+LPS group, as compared to the IgG+LPS group. In addition, EphA2 antagonism reduced the expression of phospho-p85, phosphoinositide 3-kinase 110γ, phospho-Akt, nuclear factor κB, and proinflammatory cytokines.
Conclusion
This results of the study indicated a role for EphA2-ephrinA1 signaling in the pathogenesis of LPS-induced lung injury. Furthermore, EphA2 antagonism inhibits the phosphoinositide 3-kinase-Akt pathway and attenuates inflammation.
Introduction
The Eph tyrosine kinase receptor and ephrin ligand are cell surface-bound and are involved in cell-to-cell communication12. The influence of Ephephrin activation differs depending on the cell type and environment. Eph-ephrin signaling contributes to several functions including vasculogenesis, angiogenesis, cell migration, axon guidance, fluid homeostasis and repair after injury123. A great deal of recent research has focused on the complex role of Eph and ephrin in malignancy45. According to several studies, Eph receptors and ephrin ligands affect multiple oncogenic signaling pathways such as MAPK/ERK, phosphoinositide 3-kinase (PI3K), E-cadherin and integrin/FAK/paxillin 4678. In addition to bidirectional signaling, Eph receptors and ephrin ligands function independently of each other or in conjunction with other cell surface communication systems1.
Eph-Ephrin proteins may play a role in inflammation through phenotypic changes in the vascular endothelium, which allow movement of inflammatory cells into the injured tissue3. There are studies on the role of EphA2-ephrinA1 signaling in lung injury91011. These studies have shown that both EphA2 and ephrinA1 increase in bleomycin-induced lung injury and lung injury due to viral infection and hypoxia910, contrary to counter-directed changes in expressional regulation of Eph and ephrin during lipopolysaccharide (LPS) fever in rats11. In the bleomycin-induced lung injury model, EphA2 knockout mice showed reduced permeability and less inflammatory response than wild type mice9. Similarly, in lung injury due to viral infection and hypoxia, EphA2 antagonism with EphA2/Fc and anti-EphA2 antibody reduced vascular leakage and albumin extravasation10.
In this study, we investigated the expression of Eph-ephrin proteins and the effect of EphA2 monoclonal antibody (mAb) pretreatment in an LPS induced lung injury model. Also, we studied the association between EphA2 signaling and other known signal pathways of LPS-induced lung injury.
Materials and Methods
1. Experimental animals
Experiments were conducted in accordance with protocols approved by the institutional animal care committee of the Medical College of Yonsei University. Wild type male C57BL/6J mice, 8-10 weeks of age and weighing 20-24 g were purchased from Orient Bio (Seongnam, Korea). All animals were supplied with food and water and were subjected to a similar day and night light cycle.
2. Study design
Animals were studied in three experimental groups: the IgG+phosphate-buffered saline (PBS) group, the IgG+LPS group and the EphA2 mAb+LPS group. The EphA2 mAb+LPS group was divided into two subgroups according to the dose of EphA2 mAb (2 µg and 4 µg). Under inhalation anesthesia using isoflurane, the mice inhaled 4 µg of mouse IgG (Cat# ab37355; Abcam, Cambridege, MA, USA) or monoclonal rat EphA2 antibody (Cat# MAB639; R&D Systems, Minneapolis, MN, USA) in 50 µL PBS 1 hour prior to instillation experiments10. After pretreatment, mice underwent intranasal instillation of 50 µL PBS or Escherichia coli LPS (E.coli 0127: B8; Sigma, St. Louis, MO, USA) 40 µg/g in 50 µL PBS. The intranasal administration was performed by gradually releasing the solution into the nostril with the help of microsyringe (Cat# 7637-01; Hamilton Company, Reno, NV, USA)12. The rate of release was adjusted to allow the mouse to inhale the solution completely without trying to form bubbles.
All mice were sacrificed 48 hours after treatment and lung tissue was harvested for analysis. For bronchoalveolar lavage fluid (BALF) studies, lungs were lavaged via the trachea with two 1-mL aliquots of saline. After the BALF was centrifuged, the cell pellets were used for differential cell counts and the supernatants were used for quantitative analysis of protein by the Bradford protein assay. While left lung tissue was used for histological studies, right lung tissue was prepared for Western blotting and quantitative cytokine enzyme-linked immunosorbent assay (ELISA).
3. Histopathology
Left lungs were inflated and fixed with 4% paraformaldehyde before being embedded in paraffin and sectioned. Semiquantitative grading of lung injury on hematoxylin and eosin-stained sections was performed as previously described13.
4. Western blotting and cytokine ELISA
The right lung homogenate lysates were centrifuged at 13,000 ×g for 30 minutes at 4℃. Equal amounts of protein were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane before immunoblotting with primary antibodies, as indicated. Membranes were incubated with anti-rabbit or anti-mouse IgG horseradish peroxidase conjugated antibodies and developed using a Super-Signal West Pico chemiluminescence detection kit (Pierce, Rockford, IL, USA). The band images were quantified using the Alpha Ease FC software version 4.1.0 (Alpha Innotech, San Leandro, CA, USA).
The antibodies used in this study included rabbit EphA2 (1:1,000, Thermo Fisher Scientific, Bremen, Germany), goat ephrinA1 (1:1,000, Thermo Fisher Scientific), rabbit phosphate-Akt (1:1,000, Cell Signaling Technologies, Beverly, MA, USA), rabbit Akt (1:1,000, Cell Signaling Technologies), rabbit phosphate-p85 (1:500, Cell Signaling Technologies), rabbit p85 (1:1,000, Cell Signaling Technologies), rabbit PI3 kinase 110γ (1:1,000, Cell Signaling Technologies), rabbit α-tubulin (1:1,000, Cell Signaling Technologies), rabbit phosphate-p65 nuclear factor κB (NF-κB; 1:500, Thermo Fisher Scientific), rabbit p65 NF-κB (1:500, Thermo Fisher Scientific), mouse phosphate-IκBα (1:500, Cell Signaling Technologies) and mouse IκBα (1:500, Cell Signaling Technologies). Measurement of keratinocyte chemoattractant (KC/CXCL1), macrophage inflammatory protein 2 (MIP-2), interleukin (IL)-1β, IL-6, and tumor necrosis factor α (TNF-α) were performed on whole-lung homogenates using quantified ELISA kits (MILLIPLEX MAG Mouse Cytokine/Chemokine kit from Millipore, Billerica, MA, USA).
5. Statistical analysis
Statistical analysis was performed using Prism version 5.0 (Graphpad Software, Durham, NC, USA). Two group comparisons were performed with an unpaired student t test. Data are expressed as mean±SD for each group. Differences were considered significant at p<0.05.
Results
1. EphA2 and ephrinA1 are upregulated in LPS induced lung injury
As shown in Figure 1, Western blotting studies demonstrated a significant increase in EphA2 and ephrinA1 protein expression after LPS challenge. Both EphA2 and ephrinA1 protein expression were reduced when EphA2 mAb was instilled as pretreatment. Also, the degree of decline was higher in the 4 µg EphA2 mAb subgroup than the 2 µg mAb subgroup in both EphA2 and ephrinA1 (Figure 1).

The expression of EphA2 and ephrinA1 in lung lysates after lipopolysaccharide (LPS) exposure, as shown by Western blots (A) and densitometry (B, C). IgG pretreatment increased the expression of EphA2 and ephrinA1 after LPS exposure; whereas, EphA2 monoclonal antibody pretreatment reduced the expression of EphA2 and ephrinA1 after LPS exposure. Values are presented as mean±SD. PBS: phosphate buffered saline.
2. EphA2 antagonism attenuates LPS induced lung injury
In the IgG+PBS group, neutrophil recruitment, alveolar wall thickening and hemorrhage were not observed, contrary to the results of the IgG+LPS group (Figure 2A), where hyaline membrane formation, neutrophils in the alveolar and interstitium space and thickening of alveolar septa were remarkably increased (Figure 2B). The EphA2 mAb+LPS groups demonstrated reduced neutrophil infiltration and alveolar thickening compared with the IgG+LPS group (Figure 2C, D).

The effect of EphA2 monoclonal antibody pretreatment in lipopolysaccharide (LPS)-induced lung injury. Mice were intranasally treated with IgG or EphA2 antibody. One hour later, they were intranasally treated with phosphate buffered saline (PBS) or 40 µg LPS in PBS. The lungs were removed after 24 hr and stained with hematoxylin and eosin (H&E). (A) IgG+PBS group, (B) IgG+LPS group, (C) EphA2 Ab 2 µg+LPS group, and (D) EphA2 Ab 4 µg+LPS group, n=4 per group. The IgG+LPS group (B) showed obvious neutrophil infiltration and alveolar septal infiltration; however, the EphA2 Ab+LPS groups (C, D) showed attenuated lung injury (H&E stain, ×400).
Figure 3 shows the lung injury scores, bronchoalveolar lavage cell counts and protein concentrations of the four groups. The lung injury score of the EphA2 mAb 4 µg+LPS group was lower than that of the IgG+LPS group (4.30±2.93 vs.11.45±1.20, respectively; p=0.004). LPS caused a significant increase in cell counts and concentration of total protein in the lung lavage fluid. LPS-induced lung injury was attenuated by pretreatment with EphA2 mAb. Cell counts were significantly less (EphA2 mAb 4 µg+LPS: 11.33×104±8.84×104 vs. IgG+LPS: 208.0×104±122.6×104; p=0.018) and concentrations of total protein were lower (EphA2 mAb 4 µg+LPS: 0.52±0.41 mg/mL vs. IgG+LPS: 1.38±1.08 mg/mL; p=0.192) in EphA2 mAb+LPS group, compared with IgG+LPS group.

The effect of EphA2 monoclonal antibody pretreatment on semiquantitative lung injury scores (A), total bronchoalveolar fluid (BALF) cell count (B) and BALF protein concentration (C). In the lipopolysaccharide (LPS)-induced lung injury model, EphA2 antagonism with EphA2 monoclonal antibody led to improved lung injury scores, decreased BALF cell counts and decreased BALF proteins (p<0.05, except BALF protein), as compared to IgG pretreatment. Values are presented as mean±SD (n=4 per group).
3. EphA2 regulates expression of the PI3K-Akt pathway
To elucidate whether the LPS-induced injury related signal pathway is regulated by EphA2-ephrinA1 signaling, the PI3K-Akt pathway was assessed by Western blot analysis in the IgG+PBS, IgG+LPS, and EphA2 mAb+LPS groups (Figures 4, 5B).
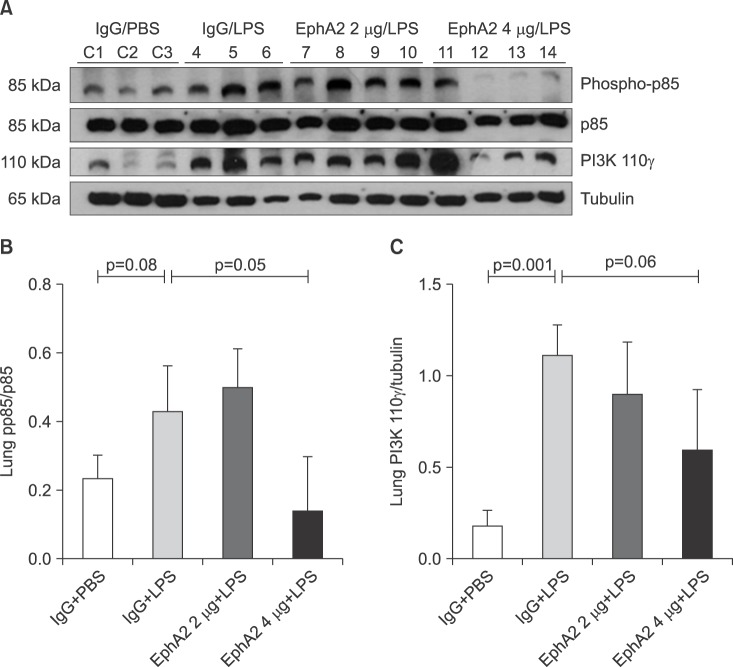
The expression of phosphoinositide 3-kinase (PI3K) protein (pp85 and 110γ) in lung lysates after lipopolysaccharide (LPS) exposure, as shown by Western blots (A) and densitometry (B, C). Phosphorylated 110γ (catalytic subunit of PI3K) levels were increased in mice with LPS exposure (IgG+LPS), as compared with controls (IgG+phosphate buffered saline [PBS]). Activation of P110γ was reduced in response to EphA2 monoclonal antibody pretreatment (EphA2+LPS) (C). However, the expression of p85 (regulatory subunit of PI3K) protein was not consistently influenced by EphA2 monoclonal antibody (B). Values are presented as mean±SD.
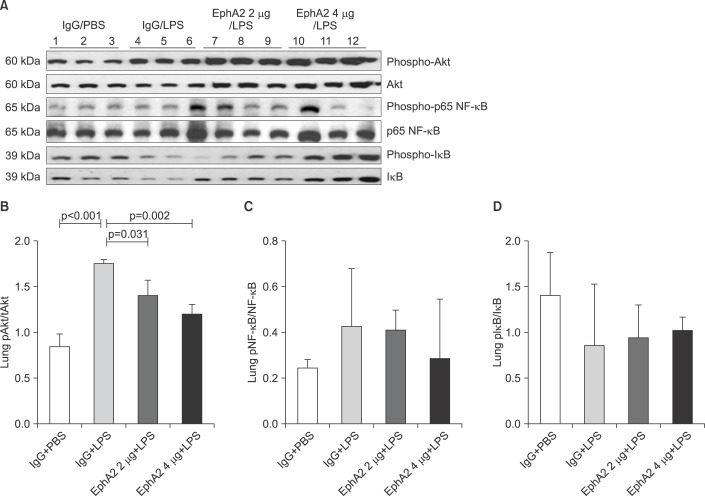
The expression of Akt, p65 nuclear factor κB (NF-κB), and IκB in lung lysates after lipopolysaccharide (LPS) exposure, as shown by representative Western blots (A) and densitometry (B-D). The amount of phosphorylated Akt was increased in mice with LPS exposure (IgG+LPS), as compared with controls (IgG+PBS). LPS-induced activation of Akt was reduced in response to EphA2 monoclonal antibody pretreatment (EphA2+LPS) (B). The expression of phosphorylated NF-κB protein was similar to that of Akt (C). The expression of phosphorylated IκB after LPS exposure was increased in mice with EphA2 monoclonal antibody pretreatment (D). Values are presented as mean±SD.
Compared to the IgG+PBS group, the IgG+LPS group showed significantly increased phospho-p85 and PI3K 110γ and phospho-Akt. After EphA2 mAb pretreatment, the expression of PI3K 110γ and phospho-Akt by LPS challenge were reduced, which indicated the inhibitory effect of EphA2 mAb (PI3K 110γ, p=0.06; p-Akt, p=0.002). The regulatory subunit PI3K phospho-p85 was not influenced by EphA2 mAb pretreatment.
4. EphA2 antagonism decreased NF-κB expression and cytokine production
To determine whether EphA2 mAb has a protective function in inflammatory response, we assessed the expression of p65 NF-κB, IκB protein and cytokine levels (IL-1β, IL-6, KC, MIP-2, and TNF-α) by Western blot analysis and ELISA. EphA2 antagonism led to a decrease of p65 NF-κB and cytokine production in the EphA2 mAb+LPS group compared to the IgG+LPS group (Figures 5B, 5C, 6).
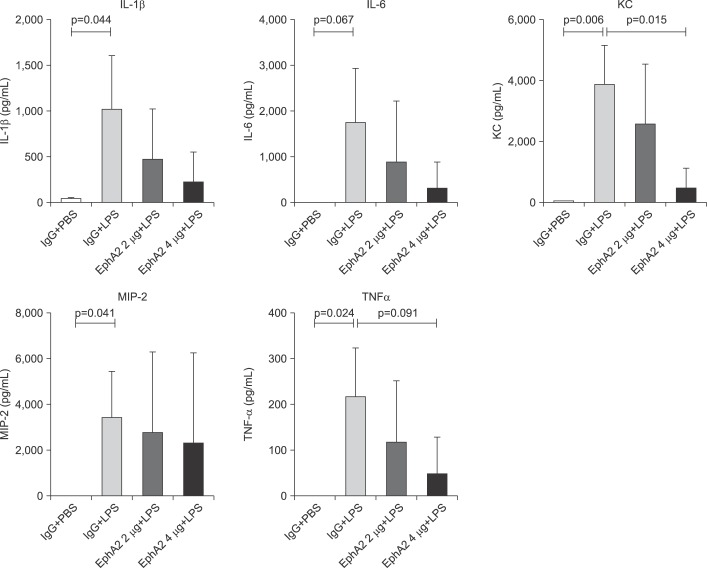
The expression of cytokines (interleukin [IL]-1β, IL-6, KC, macrophage inflammatory protein 2 [MIP-2], and tumor necrosis factor α [TNF-α]) in response to EphA2 monoclonal antibody pretreatment in a lipopolysaccharide (LPS) induced lung injury model. Enzyme linked immunosorbent assay measurement showed differential expression of cytokines (IL-1β, IL-6, KC, MIP-2, and TNF-α) in lung tissue lysates. Cytokine levels were reduced in response to LPS exposure in mice with EphA2 monoclonal antibody pretreatment, as compared to those with IgG pretreatment (n=3 per group). Values are presented as mean±SD.
5. EphA2-ephrinA1 signaling in LPS-induced lung injury
EphrinA1-dependent EphA2 activation may be an unrecognized contributor to LPS-induced lung injury. Antagonizing EphA2 not only resulted in diminished expression of the PI3Kγ isoform, but it also led to decreased NF-κB activation and expression of proinflammatory cytokines through the PI3K-Akt pathway. Crosstalk of EphA2 signaling with the PI3K-Akt pathway may contribute to the neutrophil-driven inflammatory process in LPS-induced lung injury (Figure 7).

The effects of ephrinA1-mediated activation of EphA2 on the phosphoinositide 3-kinase (PI3K)/Akt signaling pathways in lipopolysaccharide (LPS)-induced lung injury. EphA2-ephrinA1 signaling may contribute to the development of LPS-induced lung injury. Exposure to LPS resulted in activation of the EphA2-ephrinA1 and PI3K/Akt/nuclear factor κB (NF-κB) signaling pathway. EphA2 monoclonal antibody-induced inhibition of EphA2 signaling resulted in decreased PI3K/Akt/NF-κB-dependent inflammatory processes. These results suggested crosstalk between EphA2 signaling and PI3K/Akt/NF-κB signaling in LPS-induced lung injury. TNF-α: tumor necrosis factor α; IL: interleukin.
Discussion
Although acute lung injury accounts for significant morbidity and mortality in critically ill patients, there are no specific effective therapies14. Thus, it is important to find a therapeutic target and verify the effectiveness of related treatment in acute lung injury.
This study demonstrates that the expression of the EphA2 receptor and ephrinA1 ligand is upregulated in an LPS-induced lung injury model. Blockage of EphA2 activation leads to reduction of protein leakage, recruitment of inflammatory cells and cytokine production. Furthermore, the EphA2 receptor is involved in the PI3K/Akt signal pathway in LPS-induced NF-κB activation.
Eph receptors and ephrin ligands represent the largest family of receptor tyrosine kinases that regulate important processes during embryonic neuronal development, angiogenesis and oncogenesis12. Emerging evidence suggests that Eph/ephrin plays a key role in inflammation3 and previous studies have established the role of EphA2 in postnatal lung injury 9101115.
Contrary to our results that both EphA2 and ephrinA1 increased after LPS exposure, Ivanov et al.11 demonstrated opposing changes in expressional regulation of the EphA2 receptor and ephrin ligand in lung tissue in phase 2 (90 minute post-LPS) of LPS exposure. The difference between the two studies may be due to the experiment time interval. The sacrifice time was 90 minutes after LPS exposure in the study of Ivanov et al.11, whereas it was 48 hours after LPS exposure in our study.
Our results are consistent with previous studies on lung injury after instillation of bleomycin and exposure to hypoxia after viral infection910. Both studies showed increased expression of EphA2 and ephrinA1 in lung injury and found that EphA2 receptor antagonism (via a EphA2 KO mouse model or EphA2 mAb pretreatment) provided protection against lung injury showing reduced permeability and protein extravasation.
EphA2 contributes not just to actin cytoskeleton disruption leading to vascular permeability, but also to inflammation in lung injury. Several data suggest that actin cytoskeleton rearrangement may be a key preceding event during the regulation of inflammatory responses161718. Our results have revealed that EphA2 mAb pretreatment ameliorates lung injury by attenuating the expression of cytokines and neutrophil infiltration. This is similar to bleomycin-induced lung injury, where the proinflammatory transcriptional factors NF-κB, CXCL1, and CCL2 are triggered by EphA2 stimulation via the ephrin ligand in wild mice, but inhibited in EphA2 deficient mice9.
Although the main effect of Eph receptor-ephrin ligand signaling is increased vascular permeability and inflammatory cell migration, the detailed mechanism in lung inflammation remains unclear.
PI3K and Akt occupy an important role in modulation of NF-κB activation and proinflammatory cytokine production in numerous cell populations including neutrophils, epithelial cells and fibroblasts19202122. Yum et al.23 showed that, after exposure to LPS, Akt is activated via PI3Kγ and this leads to enhanced nuclear translocation of NF-κB and reduced lung neutrophil apoptosis. The present study demonstrated that antagonizing EphA2 expression with EphA2 mAb blocked the PI3K-Akt-NF-κB pathway. Another study supported a direct correlation between EphA2 signaling and the PI3K-Akt pathway. Holen et al.24 reported that reverse signaling through the ephrin A4 ligand by interaction with the EphA2 receptor led to Src kinase phosphorylation, PI3K-Akt activation and inhibition of cell death by apoptosis.
In view of this, the EphA2 receptor may be an important mediator for crosstalk between the inflammatory signal pathway and the cell-to-cell integrity pathway. Considering that the expression of ephrinA1 is induced by some cytokines2526, ligand-mediated activation of the EphA2 receptor is expected to be accelerated in lung injury. Recent research reported that EphA2 expression induced by tobacco smoke exposure in bronchial airway epithelial cells was dependent on MAPK (ERK, p38, and JNK) signaling15. Determining whether EphA2 signaling communicates with these additional mechanisms in LPS-induced lung inflammation will require further investigation.
Since EphA2-ephrinA1 signaling was reported to regulate multiple oncogenesis events27, clinical trials of drugs inhibiting the EphA2 receptor have been ongoing in cancer patients2829. Similarly, the possible role of EphA2 as a therapeutic target in this LPS-induced lung injury model should be verified in human studies.
In conclusion, we demonstrate that EphA2 signaling regulates permeability and inflammation in LPS-induced lung injury and leads to activation of the PI3K-Akt-NF-κB pathway. Moreover, the inhibition of EphA2 expression by EphA2 mAb instillation reduces neutrophil infiltration, proinflammatory cytokine production and alveolar edema. Therefore, the EphA2 receptor may present a potential therapeutic target in acute lung injury.
Acknowledgements
This Study was Supported by a 2013-Grant from The Korean Academy of Tuberculosis and Respiratory Diseases.
Notes
Conflicts of Interest: No potential conflict of interest relevant to this article was reported.