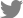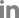Introduction
The idiopathic inflammatory myopathies (IIM) are a rare group of heterogeneous connective tissue diseases affecting skeletal muscle and other organ systems. Subsets of IIM are clinically and pathologically assigned to one of three general subcategories: polymyositis (PM), dermatomyositis (DM), and inclusion body myositis. The lung is one of the most common extramuscular targets in IIM, and interstitial lung disease (ILD) is a prevalent and often devastating manifestation of IIM. IIM-associated ILD (IIM-ILD) contributes to nearly 80% of the mortality in IIM with a reported prevalence of 65% of newly diagnosed IIM cases1,2,3. The antisynthetase syndrome is characterized by the presence of any one of several antisynthetase autoantibodies (perhaps occurring in up to one-third of DM and PM patients) in combination with fever, Raynaud's phenomenon, polyarthritis, myositis, ILD, and "mechanic hands"4,5. ILD often dominates the clinical picture and may be the presenting feature of the syndrome, as the acute onset of fever and respiratory distress occurs in nearly 50% of patients3,6,7. Acute respiratory distress syndrome (ARDS) is the most acute form of presentation, with rapid progressive respiratory failure; however, very few cases have been reported in association with IIM or the antisynthetase syndrome8,9.
We report a case with antisynthetase syndrome, presenting with ARDS.
Case Report
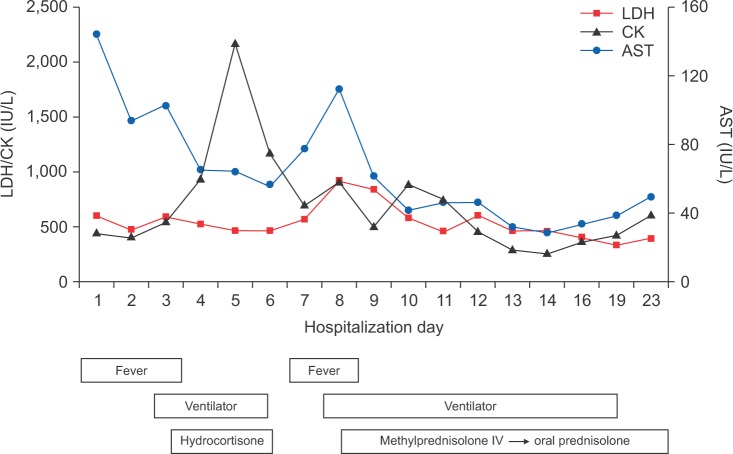
A 60-year-old woman was admitted to the Emergency Department (ED) of our hospital for cough, shortness of breath, chills, and myalgia. There was no sputum. She was a tourist from Japan and had come to Korea three days earlier. The patient had been in good health until the past week, when she developed a fever and headache. In response, she took acetaminophen for 3 days. She had no history of smoking and was not on any continuous therapy. On arrival to the ED, the patient's body temperature was 37.8Ōäā, blood pressure 130/74 mm Hg, pulse rate 82 beats per minute, respiratory rate 24 breaths per minute, and oxygen saturation 86% while breathing ambient air. Crackles were heard at the base of lungs. Respiratory failure was confirmed by the arterial blood gas: PO2 51 mm Hg, PCO2 32 mm Hg, and pH 7.52. The white blood cell count was 7,500/┬ĄL (neutrophils, 80%; lymphocytes, 12%; monocytes, 5%) and a hemoglobin value of 13.0 g/dL. C-reactive protein was increased to 12.7 mg/dL (normal, 0-0.8 mg/dL). Procalcitonin was <0.05 ng/mL (normal, <0.05 ng/mL). Chest radiography (Figure 1B) showed bilateral infiltrates in both lower fields. The patient was admitted to the Respiratory Department, where she received antibiotics (intravenous levofloxacin plus piperacillin-tazobactam plus oral clarithromycin) and oxygen. Further blood tests were conducted, which showed elevations of creatinine kinase (448 IU/L), lactate dehydrogenase (596 IU/L), aspartate aminotransferase (144 IU/L), and alanine aminotransferase (128 IU/L). On the second day of hospitalization, the patient's respiratory failure worsened, and she was transferred to the intensive care unit, where endotracheal intubation and mechanical ventilation were performed. As she progressed to respiratory failure (Figure 1C), we thought she had severe community-acquired pneumonia, and we administered hydrocortisone intravenously (100 mg then 50 mg every 6 hours) for 3 days. After 4 days on the ventilator (Figure 1D), we removed the endotracheal tube. However, two days later, the patient's body temperature was 39Ōäā, her respiratory rate was 35 breaths per minute, and she complained of general weakness and dyspnea. Due to the development of ARDS (Figure 1E), we re-intubated the patient and began mechanical ventilation again. We thought her condition may be ventilator-associated pneumonia, and changed antibiotics from levofloxacin plus piperacillin-tazobactam to meropenem plus vancomycin. However, the patient's high fever persisted and her chest radiography worsened. On the eighth day of hospitalization, there was still no improvement and the microbiological evaluation provided no evidence for bacterial, fungal, or viral infection under repeated endotracheal aspiration cultures and protected bronchial washing culture. We suspected inflammatory myopathy with ILD due to the increased muscle and liver enzyme values. Especially, creatinine kinase was elevated to >2,000 IU/L. Thus, we checked specific markers for connective tissue diseases. In addition, we reviewed her past medical history from her daughter. Before admission, the patient had suffered from frequent oral ulcers and eye dryness for several weeks. Two months ago, she underwent chest radiography (Figure 1A) at a Japanese hospital and the results were normal. One week before she arrived in Korea, she underwent a blood test due to multiple arthralgias, which showed a high value of lactate dehydrogenase (422 IU/L). While we screened for the specific markers for inflammatory myopathy, an empirical therapy with high-dose corticosteroids was started (intravenous pulses of 125 mg every 6 hours for one day tapered 60 mg/day). The therapy led to a significant improvement of PaO2/FIO2 ratio, and enabled to wean the patient from mechanical ventilation on the 11th day after intubation. On the 13th day of hospitalization, laboratory results revealed positive anti-Jo-1 antibody with 6.4 index (normal range, <1.0 index), aldolase with 22.9 U/L (normal range, 0-7.6 U/L), and anti-Ro (SSA) antibody with >240 U/mL (normal range, <10 U/mL). Rheumatoid factor, antinuclear antibody, anti-La (SSB) antibody, anti-centromere antibody, anti-Scl-70 antibody, anti-ds-DNA antibody, anti-ribonucleoprotein antibody, and anti-Sm antibody were all negative. On high-resolution computed tomography (HRCT) (Figure 2) after extubation, there were still irregular diffuse ground-glass opacity in both upper lungs and irregular patchy consolidation on both lower lungs without honeycombing. Although a histologic diagnosis was not made, we provisionally diagnosed the patient with ARDS as the initial clinical manifestation of an anti-Jo-1 antibody-positive antisynthetase syndrome. After 25 days of hospitalization (Figure 1F), the patient was discharged without oxygen. Figure 3 shows the whole course of laboratory values, clinical course, and treatments.
Discussion
Aminoacyl-tRNA synthetase (ARS) is a family of cytoplasmic enzymes that catalyze the formation of aminoacyl-tRNA from a specific amino acid and its cognate tRNA and play a crucial role in protein synthesis. Eight anti-ARS autoantibodies have been identified to date. These include antihistidyl (anti-Jo-1), anti-threonyl (anti-PL-7), anti-alanyl (anti-PL-12), anti-isoleucyl (anti-OJ), anti-glycyl (anti-EJ), anti-asparaginyl (anti-KS), anti-phenylalanyl (anti-Zo), and anti-tyrosyl-tRNA synthetases7. Based on a unique combination of clinical features commonly observed in patients with anti-ARS autoantibodies, Targoff4 proposed a disease entity termed antisynthetase syndrome. Antihistidyl tRNA synthetase antibody, often referred to as anti-Jo-1 antibody, is the most frequently detected antisynthetase autoantibody and is strongly associated with the presence of ILD in both PM and DM. In one large cohort study, ILD was identified in 86% of anti-Jo-1 antibody-positive myositis patients10.
The breadth of presentation in IIM-ILD ranges from asymptomatic basilar lung fibrosis to an acute, rapidly progressive condition associated with ARDS and respiratory failure3,6. The most common presenting symptoms are cough and dyspnea; however, IIM-ILD may be associated with asymptomatic lung disease, as evidenced by the high frequency of radiographically documented ILD10. Of clinical importance, there is no correlation between the severity of lung involvement and the degree of myositis disease activity; yet, the severity of respiratory symptoms at presentation appears to correlate with the outcome in IIM-ILD11,12. In particular, acute and rapidly progressive ILD, corresponding to ARDS with the histopathology of diffuse alveolar damage (DAD), can occur in any patient with IIM-ILD but is frequently seen with the antisynthetase syndrome, regardless of whether patients manifest complete forms of the syndrome or formes frustes4.
Several histopathologic patterns have been described in IIM-ILD3,13. The most common histology in IIM-ILD is nonspecific interstitial pneumonia (NSIP), with a prevalence of approximately 80%3. Usual interstitial pneumonia is less common than NSIP in IIM-ILD3. DAD is the most rapidly progressive pathologic subset and carries worst prognosis. DAD, resembling the histological appearance associated with ARDS, is diffuse interstitial inflammation with edema and hyaline membrane production ultimately leading to organizing fibrosis. On HRCT, it is diffuse in appearance with ground glass consolidations and, in later stages, traction bronchiectasis and honeycombing may appear. Any initial HRCT pattern can rapidly deteriorate to ARDS, often leading to patient demise3. In one retrospective case study, ILD patients with anti-ARS antibodies showed a chronic clinical course, lung-base predominant ground glass opacity with volume loss, NSIP and/or organizing pneumonia patterns, and a good response to corticosteroid treatment; however, some had a rapidly worsening courses and recurrence, despite therapy7. In terms of prognosis, Yousem et al.13, studying the clinicopathologic pattern of 20 antisynthetase syndrome patients, found that the subgroup of patients presenting with an acute rapidly progressive hypoxemia had a worst prognostic histologic pattern due to the presence of DAD in five cases. This analysis highlights that a significant portion of these patients present with acute respiratory decompensation manifesting as pure DAD or DAD superimposed on underlying chronic symptomatic or asymptomatic fibrosing lung disease13. Our patient followed a fulminant course, with fever, rapid progression of radiographic changes, and respiratory failure. Her clinical course was all consistent with acute interstitial pneumonia, although we did not obtain a surgical lung biopsy.
There are no approved agents in the treatment of IIM, and this lack of established treatment regimen extends to IIM-ILD. Nevertheless, corticosteroids remain the cornerstone of early treatment with initial doses at 1 mg/kg of the ideal body weight. In an effort to reduce treatment-related side effects, other immunosuppressive, steroid-sparing agents should be considered at the outset of therapy, particularly, when treating antisynthetase syndrome and other severe and progressive manifestations of ILD3. Especially, given the relative rarity of ARDS complicating IIM, there is a paucity of studies to guide therapy8,14. Perhaps the best approach for now is to increase the intensity of pulse cyclophosphamide, cyclosporine, or other immunosuppressive therapy earlier, especially in patients who have responded poorly, if at all, to conventional pulse steroid therapy. Tacrolimus, 100-fold more potent than cyclosporine in inhibiting T-cell activation, has also been used in concert with other immunosuppressive agents15. Fortunately, our patient's ARDS secondary to antisynthetase syndrome was resolved under treatment with a high dose of steroids, resulting in the patient's discharge without oxygen.
In conclusion, given that steroids are not greatly beneficial in the treatment of ARDS, it is likely that the improvement of the respiratory symptoms in this patient also resulted from the prompt suppression of the inflammatory systemic response by corticosteroid.
 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Print
Print Download Citation
Download Citation