Introduction
Lung cancer is one of the most commonly diagnosed cancers and the leading cause of cancer-related deaths worldwide1. In Korea, lung cancer was the fourth most common cancer in 2013, with 23,177 newly diagnosed patients, and the most common cause of cancer-related deaths, accounting for 22.8% of all cancer-related deaths in 20142,3. Non-small cell lung cancer (NSCLC) is diagnosed in approximately 85% of patients with lung cancer, but most of these patients are diagnosed at an inoperable or advanced stage4.
Progress in the treatment of advanced NSCLC over the past decade includes the introduction of the cytotoxic chemotherapy agent pemetrexed for tumors of non-squamous histology and the development of molecularly targeted agents, including epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKI) and anaplastic lymphoma kinase (ALK) inhibitors, for tumors with activating mutations, most of adenocarcinoma histology4,5. These agents have resulted in better survival outcomes4,5. The frequency of EGFR mutations in tumors of adenocarcinoma is highest in patients from Asia-Pacific countries, at 47% (range, 20%-76%)6. Moreover, the incidence of adenocarcinoma is steadily increasing, making it the most common subtype of lung cancer in Korea7.
These advanced treatments, however, are unavailable to patients with squamous cell histology and those without appropriate molecular alterations, making their prognosis as poor as ever. Moreover, acquired resistance to targeted agents is now a challenging problem in patients who progress on these therapies4,5. Therefore, the 5-year survival rate in patients with lung cancer ranges remains only 10%-20% in both developed and developing countries, despite improving up to 10% in most countries8. Obviously, new therapeutic options are required for patients with advanced NSCLC and unmet medical needs.
Immunotherapy, which uses a patient's own immune system, has recently appeared as another modality for cancer treatment. Immuno-oncology has become an important focus of basic research and clinical trials for the treatment of NSCLC9,10,11,12,13,14,15. This review summarizes basic tumor immunology and clinical data on immunotherapeutic approaches, especially immune checkpoint inhibitors in NSCLC.
Immune System and Tumor Immunology
Historically, immunity has signified a defense mechanism against infectious diseases. However, noninfectious foreign substances can also elicit immune responses. Therefore, the immune system reacts not only to infectious microbes but to cancer cells, and has the potential to kill cancer cells16.
The immune system consists of the innate immune system, which reacts initially to foreign substances, and the adaptive immune system, which responds subsequently. The innate immune system includes complement proteins and cellular components, including natural killer cells (NKs), dendritic cells (DCs), polymorphonuclear leukocytes, mast cells, and macrophages. The adaptive immune system includes humoral immunity mediated by antibodies produced by B lymphocytes, and cellular immunity mediated by T lymphocytes16,17,18. Natural killer T (NKT) cells and γδ T cells are involved in both innate and adaptive immunity17. The innate immune system is ready to respond rapidly, in the absence of prior exposure, and is antigen-nonspecific. By contrast, the adaptive immune response is slower to develop, educated to recall prior exposure, known as memory, and is antigen-specific16,17,18.
Cancer immunotherapy is an evolving treatment modality that uses a patient's own immune system to fight cancer. Theoretically, cancer immunotherapy can result in long-term cancer remission and may not cause the same side effects as chemotherapy and radiation19,20.
Classically, cancer immunosurveillance hypothesizes that the immune system can recognize and eliminate nascent transformed cells21. However, tumors that developed in immunocompetent mice were found to be less immunogenic than tumors that developed in immunodeficient mice22. These findings indicated that, paradoxically, the immune system assists in the eventual outgrowth of cancers that are better able to escape immune detection. Thus, tumors are imprinted by the immunologic environment in which they form22,23.
1. Cancer immunoediting
Because the immune system can promote as well as suppress cancer growth, the broader term cancer immunoediting was proposed to describe better these activities, in place of the term cancer immunosurveillance22,23,24.
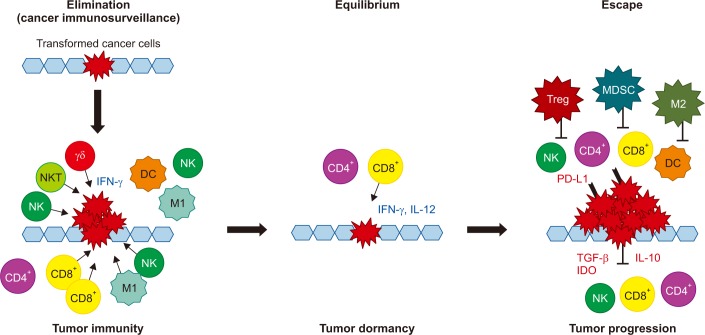
Cancer immunoediting consists of three phases of relations between cancer cells and the immune system: elimination, equilibrium, and escape (Figure 1). In the elimination phase, the immune system detects and destroys transformed cancer cells from normal tissue before they become clinically detectable tumors. This phase corresponds to the concept of cancer immunosurveillance, in which the innate and adaptive immune systems work together. A proposed model for the elimination phase consists of four steps. In the first step, innate immune cells, such as NKs, NKT cells, and γδ T cells, recognize transformed cells and produce interferon γ (IFN-γ). In the second step, IFN-γ induces the death of a limited number of tumor cells and some chemokines recruit NKs, DCs, and macrophages. DCs ingest dead tumor cells and migrate to the draining lymph node. In the third step, NK cells and macrophages produce interleukin (IL)-12 and IFN-γ, which kill additional tumor cells while tumor antigen-specific T cells develop in the draining lymph nodes. In the fourth step, tumor antigen-specific T cells home to the tumor site and destroy tumor cells23,24.
During the equilibrium phase, any cancer cell variant that has survived the elimination phase has increased resistance to immune recognition. The immune system also holds cancer cells in a functionally dormant state using adaptive immune cells such as T cells. Thus, the immune system prevents cancer cell outgrowth and sculpts the immunogenicity of these cancer cells23,24.
During the escape phase, the immune system can no longer block tumor cells outgrowth, resulting in the emergence of cancer cells and the development of clinically observable malignant disease. Cancer cell escape can occur through many different mechanisms, including the loss of tumor antigens, resulting in the evasion of immune recognition, increased resistance to the cytotoxic effects of the immune system, and establishment of an immunosuppressive tumor microenvironment23,24.
2. Cancer-immunity cycle
An anticancer immune response can be divided into several steps, known as the cancer-immunity cycle (Figure 2)25. First, tumors release tumor cell antigens, which are captured by antigen presenting cells (APCs) such as DCs. The APCs subsequently process and present these captured antigens on major histocompatibility complex (MHC) molecules, and migrate to draining lymph nodes. In the lymph nodes, T cell receptors on the surface of T cells recognize the antigenic peptides presented by the MHC. However, T-cell activation also requires the interaction of co-stimulatory signals, such as between proteins of the B7 family (CD80 or CD86) on APCs and CD28 on T cells. Finally, activated T cells migrate to and infiltrate the tumor bed, binding to tumor cells and killing them25,26.
Each step of the cancer-immunity cycle may be a potential target for immunotherapy. Vaccines can promote tumor antigen presentation by APCs, cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) blockade can promote the priming phase, and programmed cell death protein 1 (PD-1) blockade or programmed death ligand 1 (PD-L1) blockade can promote the effector phase, consisting of the killing of cancer cells by activated T cells25,26.
3. Immunosuppressive mechanism
After T cells are activated, a checkpoint system is triggered to inhibit further T-cell activation. Originally this mechanism helps to regulate immune responses and maintain immune balance, thereby preventing autoimmune reactions. Tumors can also protect themselves from the immune system using checkpoints as immune resistance mechanisms9,15,27.
Cancer cells can induce and recruit immunosuppressive cells in the tumor microenvironment. These immunosuppressive cells include regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and M2 tumor-associated macrophages (TAMs)9,15,28. Immunosuppressive molecules such as IL-10, transforming growth factor β, and indoleamine 2,3-dioxygenase (IDO) are also secreted by cancer cells or immunosuppressive cells15,28.
4. Immune response in NSCLC
Although lung cancer has been regarded classically as a non-immunogenic malignancy, recent understanding of the immune system suggests that antitumor immune responses to lung cancer can be induced, that their magnitude may correlate with clinical outcomes, and that immunotherapy may be a therapeutic option in lung cancer9,11,15.
Several retrospective, immunohistochemical (IHC) analyses of lung cancer specimens have shown that cellular immune responses may be associated with clinical prognosis. For example, the survival rate of patients with squamous cell carcinoma of the lung, particularly those with early stage tumors, was found to be significantly higher in the presence than in the absence of tumor-infiltrating lymphocytes (TILs), which were almost entirely CD8+ T cells29. High levels of infiltration of both CD8+ T cells and CD4+ T cells were also associated with significantly higher survival rates in patients with NSCLC30. By contrast, a high ratio of Tregs to TILs correlated with a significantly higher risk of recurrence in patients with stage I NSCLC31.
Immunotherapy for NSCLC
The goal of immunotherapy is to potentiate the immune system's response to cancer cells. The adaptive immune system, especially T cells, plays an important role in anticancer immune responses15. Studies to date have focused on two immunotherapeutic strategies in NSCLC: cancer vaccines and immune checkpoint inhibitors. Cancer vaccines are antigen-specific immunotherapies that augment tumor recognition by the immune system, whereas immune checkpoint inhibitors are antigen non-specific therapies that overcome tumor immunosuppression9,11,12,15.
Vaccines
Cancer vaccines stimulate the immune system to recognize tumor antigens. Injection of a cancer vaccine, composed of tumor-associated antigens (TAA) and adjuvant, into the skin (usually intradermally) results in the uptake of TAAs by APCs such as DCs. These APCs process these TAAs and migrate to draining lymph nodes, where the activated APCs present antigens to T cells, resulting in their activation9,15,32.
Several cancer vaccines have been studied in NSCLC, including melanoma-associated antigen-A3 (MAGE-A3)33,34, liposomal-BLP25 (tecemotide)35,36, TG401037, and CIMAvax-EGF38. To date, however, these cancer vaccines remain suboptimal because the induction of the desired immune response is weak, responses are short lived, and memory formation is defective. The tumor microenvironment is also potently immunosuppressive, resulting in the rapid inactivation of vaccine-induced effector lymphocytes. These hurdles must be overcome to develop clinically effective cancer vaccines39,40. Consistently, however, these vaccines have been found more effective in patients with minimal disease burden. Patient selection based on predictive biomarkers will be a major future challenge in the development of cancer vaccines12,41,42.
Immune Checkpoint Inhibitors
Immune checkpoint inhibitors are the most promising approach for cancer immunotherapy. Immune checkpoint inhibitors most investigated in clinical trials of patients with NSCLC include antibodies against CTLA-4, PD-1, and PD-L1. A major advantage of these immune checkpoint inhibitors is their ability to elicit antitumor immune responses regardless of the specific tumor antigens12,13,14,15,27.
CTLA-4 is expressed on T cells and initiates inhibitory regulation during the priming phase of a T-cell activation. CTLA-4 competes with T-cell co-stimulatory receptor CD28, which is needed for T-cell activation, by binding to members of the B7 family (CD80 or CD86) on APCs. Binding of CTLA-4 to CD80 or CD86, rather than to CD28, provides an inhibitory signal to the T cell. By contrast, PD-1 is expressed on activated T cells and regulates the effector phase of T-cell responses in the tumor microenvironment. PD-1 binds to one of its ligands, PD-L1 or PD-L2, which is usually expressed on cancer cells, providing an inhibitory signal to the T cell. Therefore, inhibition of CTLA-4, PD-1, or PD-L1 results in the activation of T cells and enhancement of anticancer immune responses (Figure 3)12,13,14,15,27. Immune checkpoint inhibitors for the treatment of NSCLC and the results of clinical trials are summarized in Tables 1 and 2.
1. Anti-PD-1
1) Nivolumab
Nivolumab is the first anti-PD-1 antibody in clinical development. Two phase III trials compared nivolumab with docetaxel in patients with stage IIIB or IV squamous and nonsquamous NSCLC who experienced disease progression during or after one prior platinum-based chemotherapy regimen43,44. In the first phase III trial, CheckMate 017, 272 patients with squamous NSCLC were randomly assigned to receive nivolumab (3 mg/kg every 2 weeks) or docetaxel (75 mg/m2 every 3 weeks). Median overall survival (OS, 9.2 months vs. 6.0 months; hazard ratio [HR], 0.59; 95% confidence interval [CI], 0.44-0.79; p<0.001) and median progression-free survival (PFS, 3.5 months vs. 2.8 months; HR, 0.62; 95% CI, 0.47-0.81; p<0.001) were significantly longer in the nivolumab than in the docetaxel group. In addition, the 1-year OS rate (42% vs. 24%) and the objective response rate (ORR, 20% vs. 9%; p=0.008) were higher in patients treated with nivolumab than with docetaxel. Across the prespecified expression levels (1%, 5%, and 10%), the expression of the ligand for PD-1 (PD-L1) was neither prognostic nor predictive of any of the efficacy end points. Treatment-related adverse events (AEs) of grades 3 and 4 occurred less frequently with nivolumab than with docetaxel (7% vs. 55%)43.
In the second phase III trial, CheckMate 057, 582 patients with non-squamous NSCLC were randomly assigned to receive nivolumab or docetaxel, in a design similar to that of the CheckMate 017 trial. Patients with known EGFR mutation or ALK translocation were allowed to receive additional TKI therapy. Nivolumab was associated with significantly longer median OS (OS, 12.2 months vs. 9.4 months; HR, 0.73; 95% CI, 0.59-0.89; p=0.002) and higher ORR (19% vs. 12%, p=0.02). Although median PFS was shorter in the nivolumab than in the docetaxel group (median PFS, 2.3 months vs. 4.2 months), the difference was not statistically significant (HR, 0.92; 95% CI, 0.77-1.11; p<0.39). In contrast to the CheckMate 017 trial, involving patients with squamous cell NSCLC, the CheckMate 057 trial found that PD-L1 protein expression was associated with improved OS and PFS in patients treated with nivolumab. However, PD-L1 protein expression was evaluated retrospectively in prospectively collected, pretreatment (archival or recent) tumor-biopsy specimens. Grade 3 and 4 treatment-related AEs were again less frequent in patients treated with nivolumab than with docetaxel (10% vs. 54%)44.
The phase III CheckMate 026 trial compared nivolumab with platinum-based agents as first-line therapy in patients with stage IV or recurrent NSCLC. Among the 423 patients with a PD-L1 expression level ≥5%, nivolumab did not improve median PFS compared with chemotherapy (PFS, 4.2 months vs. 5.9 months; HR, 1.15; 95% CI, 0.91-1.45; p=0.25). Median OS (OS, 14.4 months vs. 13.2 months; HR,1.02; 95% CI, 0.80-1.30) and ORR (26% vs. 33%) were similar in the nivolumab and chemotherapy groups. Rates of grades 3 and 4 treatment-related AEs were lower with nivolumab than with chemotherapy (18% vs. 51%)45.
2) Pembrolizumab
The phase I KEYNOTE-001 trial found that higher PD-L1 expression in at least 50% of tumor cells (proportion score [PS] ≥50%) was associated with higher ORR and longer PFS and OS than lower levels (PS <1% and 1%-49%) in patients with advanced NSCLC46. In the subsequent phase II/III KEYNOTE-010 trial, 1,034 patients with previously treated NSCLC and PD-L1 expression (PS ≥1%) were randomly assigned to receive pembrolizumab (2 mg/kg or 10 mg/kg) or docetaxel (75 mg/m2) every 3 weeks. In the total population (PS ≥1%), median OS was significantly longer with pembrolizumab 2 mg/kg (10.4 months vs. 8.5 months; HR, 0.71; 95% CI, 0.58-0.88; p=0.0008) and with pembrolizumab 10 mg/kg (12.7 months vs. 8.5 months; HR, 0.61; 95% CI, 0.49-0.75; p<0.0001) than with docetaxel. Among patients with high PDL1 expression (PS ≥50%), median OS was significantly longer with pembrolizumab 2 mg/kg (14.9 months vs. 8.2 months; HR, 0.54; 95% CI, 0.38-0.77; p=0.0002) and pembrolizumab 10 mg/kg (17.3 months vs. 8.2 months; HR, 0.50; 95% CI, 0.36-0.70; p<0.0001) than with docetaxel. In this latter group (PS ≥50%), median PFS was also significantly longer with pembrolizumab 2 mg/kg (5.0 months vs. 4.1 months; HR, 0.59; 95% CI, 0.44-0.78; p=0.0001) and pembrolizumab 10 mg/kg (5.2 months vs. 4.1 months; HR, 0.59; 95% CI, 0.45-0.78; p<0.0001) than with docetaxel. In the total population, however, median PFS did not differ significantly in the pembrolizumab 2 mg/kg and 10 mg/kg and docetaxel groups (3.9 months vs. 4.0 months vs. 4.0 months). ORR was significantly higher in the total population of patients treated with pembrolizumab 2 mg/kg (18% vs. 9%, p=0.0005) and pembrolizumab 10 mg/kg (18% vs. 9%, p=0.0002) than with docetaxel. Similarly, ORR in patients with high PD-L1 expression (PS ≥50%) was significantly higher following treatment with pembrolizumab 2 mg/kg (30% vs. 8%, p<0.0001) and pembrolizumab 10 mg/kg (29% vs. 8%, p<0.0001) than with docetaxel. Treatment-related AEs of grades 3 to 5 were less frequent in both pembrolizumab groups than in the docectaxel group (13% vs. 16% vs. 35%)47.
The phase III KEYNOTE-024 trial compared pembrolizumab (200 mg every 3 weeks) with platinum-based chemotherapy as first-line treatment in 305 patients with stage IV NSCLC and high PD-L1 expression (PS ≥50%). Median PFS (10.3 months vs. 6.0 months; HR, 0.50; 95% CI, 0.37-0.68; p<0.001)48 and median OS (not reached vs. 14.5 months; HR, 0.63; 95% CI, 0.46-0.88; p=0.003)49 were significantly longer with pembrolizumab than with chemotherapy. ORR was higher (44.8% vs. 27.8%) and treatment-related AEs of grades 3 to 5 were less frequent (26.6% vs. 53.3%) with pembrolizumab than with chemotherapy48.
2. Anti-PD-L1
1) Atezolizumab
In the phase II POPLAR trial, 287 patients with NSCLC who progressed on post-platinum chemotherapy were randomly assigned to receive atezolizumab 1,200 mg or docetaxel 75 mg/m2 every 3 weeks. In this study, PD-L1 expression was prospectively determined by IHC in tumor cells (TCs) and tumor-infiltrating immune cells (ICs). Median OS was significantly longer in patients treated with atezolizumab than with docetaxel (12.6 months vs. 9.7 months; HR, 0.73; 95% CI, 0.53-0.99; p=0.04). The OS benefit from atezolizumab increased with increasing PD-L1 expression on TCs and/or ICs, but OS was similar with atezolizumab and doectaxel in patients without PD-L1 expression. Median PFS was similar in patients treated with atezolizumab and docetaxel (2.7 months vs. 3.0 months; HR, 0.94; 95% CI, 0.72-1.23; p=0.645). ORR was similar in these two groups (14.6% vs. 14.7%), but the median duration of response was longer with atezolizumab than with docetaxel (14.3 months vs. 7.2 months; HR, 0.41; 95% CI, 0.18-0.96; p=0.034). Treatment-related AEs of grades 3 and 4 were less frequent in patients treated with atezolizumab than with docetaxel (11% vs. 39%)50.
In the phase III OAK trial, 850 patients with stage IIIB or IV NSCLC who had received 1-2 previous lines of cytotoxic chemotherapy, including at least one platinum-based combination regimen, were randomly assigned to receive atezolizumab 1,200 mg or docetaxel 75 mg/m2 every 3 weeks. Median OS was significantly longer with atezolizumab than with docetaxel in the intention-to-treat (ITT) (13.8 months vs. 9.6 months; HR, 0.73; 95% CI, 0.62-0.87; p=0.0003) and the TC1/2/3 or IC1/2/3 (PD-L1 expression ≥1% on TCs or ICs) (15.7 months vs. 10.3 months; HR, 0.74; 95% CI, 0.58-0.93; p=0.0102) populations. The OS benefit from atezolizumab was the highest in patients with high levels of PD-L1, TC3 or IC3 (PD-L1 expression ≥50% on TCs or ≥10% on ICs) (20.5 months vs. 8.9 months; HR, 0.41; 95% CI, 0.27-0.64; p<0.0001). However, OS was also better with atezolizumab than with docetaxel in patients with TC0 and IC0 (PD-L1 expression <1% on TCs and ICs). Median PFS was similar in patients treated with atezolizumab and docetaxel in the ITT population (2.8 months vs. 4.0 months; HR, 0.95; 95% CI, 0.82-1.10; p=0.4928) and in TC1/2/3 or IC1/2/3 populations. However, median PFS was significantly longer with atezolizumab than with docetaxel in the TC3 or IC3 population (4.2 months vs. 3.3 months; HR, 0.63; 95% CI, 0.43-0.91; p=0.0123). Although ORR was similar in the two groups in the ITT (14% vs. 13%) and TC1/2/3 or IC1/2/3 populations, ORR was higher with atezolizumab than with docetaxel in patients with TC3 or IC3 (31% vs. 11%). Rates of grades 3 and 4 treatment-related AEs were lower in patients treated with atezolizumab than with docetaxel (15% vs. 43%)51.
The phase II, single-arm trial BIRCH study included 667 patients with stage IIIB or IV NSCLC and PD-L1 expression ≥5% on TCs or ICs (TC2/3 or IC2/3) who received atezolizumab as first-line, second-line, and third-line or higher treatment. ORR in the 139 patients treated with first-line atezolizumab was 22% (31% in those with TC3 or IC3). Median OS was 23.5 months (26.9 months in those with TC3 or IC3) and the 12-month OS rate was 66.4% (61.5% in patients with TC3 or IC3). Median PFS was 5.4 months (5.6 months in those with TC3 or IC3). Grade 3 and 4 treatment-related AEs occurred in 9% of these patients53.
2) Durvalumab
In the phase II, single-arm ATLANTIC study, 333 patients with stage IIIB or IV and EGFR/ALK wild-type NSCLC who had received at least two prior lines of chemotherapy were treated with durvalumab 10 mg/kg every 2 weeks. ORRs in patients with PD-L1 expression ≥25% and ≥90% were 16.4% and 30.9%, respectively. Median OS was 10.9 months in patients with PD-L1 ≥25% and was not reached in patients with PD-L1 ≥90%. One-year OS rates in patients with PD-L1 expression ≥25% and ≥90% were 47.7% and 50.8%, respectively52.
In the phase III PACIFIC trial, 709 patients with stage III NSCLC who had not progressed after platinum-based chemoradiotherapy were randomly assigned 2:1 to receive durvalumab 10 mg/kg or placebo every 2 weeks for up to 12 months. Median PFS was significantly longer with durvalumab than with placebo (16.8 months vs. 5.6 months; HR, 0.52; 95% CI, 0.42-0.65; p<0.001) and was independent of PD-L1 expression. ORR was significantly higher with durvalumab than with placebo (28.4% vs. 16.0%, p<0.001), whereas rates of grades 3 and 4 treatment-related AEs were similar in the two groups (29.9% vs. 26.1%)54.
3. Anti-CTLA-4
1) Ipilimumab
Ipilimumab has been combined with cytotoxic chemotherapy or another immunotherapy agent in patients with NSCLC. In a phase II trial, 204 patients with stage IIIB or IV NSCLC were randomly assigned 1:1:1 to receive first-line paclitaxel and carboplatin with two different schedules (concurrent or phased arm) of ipilimumab or placebo. The concurrent arm consisted of four doses of ipilimumab plus chemotherapy followed by two doses of placebo plus chemotherapy. The phased arm consisted of two doses of placebo plus chemotherapy followed by four doses of ipilimumab plus chemotherapy. The median immune-related PFS (irPFS) was longer with phased treatment than with placebo (5.7 months vs. 4.6 months; HR, 0.72; 95% CI, 0.50-1.06; p=0.05), whereas concurrent treatment did not improve irPFS55.
By contrast, a subsequent phase III trial did not show that the combination of ipilimumab plus first-line chemotherapy enhanced median OS compared with chemotherapy alone in patients with squamous NSCLC (13.4 months vs. 12.4 months; HR, 0.91; 95% CI, 0.77-1.07; p=0.25)56.
4. Combination therapies
The combination of immune checkpoint inhibitors and conventional therapy with different mechanisms of action may have a synergistic effect and result in clinical benefit. Conventional therapies, especially targeted therapy, can lead to a rapid initial response, but most responders will later acquire resistance and develop progressive disease. Conversely, immune checkpoint inhibitors can lead to a durable response in a relatively small percentage of responders57. Conventional therapies can also modulate the immune system, thereby affecting immunotherapy57,58.
The combination of two immune checkpoint inhibitors with distinct targets, particularly anti-PD-1/anti-PD-L1 and anti-CTLA-4, may also improve response rate and survival benefit compared with monotherapy58. Many ongoing clinical trials are testing combination immunotherapies, and we look forward to successful results from phase III studies.
As part of the phase II KEYNOTE-021 trial, 123 patients with non-squamous, stage IIIB or IV, chemotherapy-naive NSCLC were randomly assigned to treatment with pembrolizumab plus carboplatin and pemetrexed or to carboplatin and pemetrexed alone. The ORR was significantly higher in patients who received pembrolizumab plus chemotherapy than chemotherapy alone (55% vs. 29%, p=0.0016). In the combination group, the ORR was 80% in patients with PS ≥50%, although there was no difference between those with PS <1% and PS ≥1% (57% vs. 54%). Median PFS was significantly longer in the combination than in the chemotherapy alone (13.0 months vs. 8.9 months; HR, 0.53; 95% CI, 0.31-0.91; p=0.010)59. The ORR was higher and PFS was longer in patients treated with pembrolizumab plus chemotherapy in this trial than in those treated with pembrolizumab alone in the KEYNOTE-024 trial. Median OS was also significantly longer in the combination group than in the chemotherapy alone group (not reached vs. 20.9 months; HR, 0.59; 95% CI, 0.34-1.05; p=0.0344)60. Treatment-related AEs of grades 3 to 5 were reported in 39% of patients in the combination group compared with 26% in the chemotherapy alone group59.
In the phase I, multicohort CheckMate 012 trial, patients with stage IIIB or IV chemotherapy-naive NSCLC were assigned to receive nivolumab alone or combination therapies. In one cohort, 56 patients were assigned to receive nivolumab 10 mg/kg plus three platinum-based doublet chemotherapy agents every 3 weeks or nivolumab 5 mg/kg plus paclitaxelcarboplatin. ORRs ranged from 33% to 47% irrespective of PD-L1 expression. The 2-year OS rate in patients treated with nivolumab 5 mg/kg plus paclitaxel-carboplatin was 62%. Treatment-related grades 3 and 4 AEs were reported in 45% of patients, and the discontinuation rate was 21%61.
In another cohort of the CheckMate 012 trial, 78 patients were randomly assigned to receive nivolumab 3 mg/kg every 2 weeks plus ipilimumab 1 mg/kg every 6 or 12 weeks. The ORRs in these groups were 38% and 47%, respectively. After pooling these two groups, ORR was found to correlate with PD-L1 expression, being 92% and 57% in patients with PD-L1 expression ≥50% and ≥1%, respectively. ORR was higher in patients treated with nivolumab plus ipilimuab in this trial than with nivolumab monotherapy in another cohort of the CheckMate 012 trial. Median PFS was longer in patients treated with ipilimumab every 12 than every 6 weeks (8.1 months vs. 3.9 months). Treatment-related grade 3 and 4 AEs in these two groups were 37% and 33%, respectively62.
In a phase Ib trial (study 006), 102 patients with immunotherapy-naive, locally advanced, or metastatic NSCLC were enrolled into the dose-escalation phase and received durvalumab and tremelimumab, a selective human IgG2 monoclonal antibody against CTLA-4. Based on safety and clinical activity, durvalumab 20 mg/kg every 4 weeks plus tremelimumab 1 mg/kg were chosen as the expansion phase doses. This dose cohort showed a manageable safety profile, with a 17% rate of grades 3 and 4 treatment-related AEs, and antitumor activity (ORR 23%) irrespective of PD-L1 status63.
5. Predictive biomarkers for immune checkpoint inhibitors
The immune checkpoint inhibitors showed durable clinical responses and enhanced long-term survival in patients who benefited from these agents. However, clinical trials of unselected patients with NSCLC showed that these agents had low ORRs, of approximately 20%, as monotherapy64. Moreover, these agents have immune-related AEs, which may be life-threatening, and their costs might result in financial burdens on patients. Therefore, biomarkers predicting the benefits of these agents are required64,65.
1) PD-L1 expression
PD-L1 expression, as determined by IHC, is the most commonly used predictive biomarker for anti-PD-1 and anti-PD-L1 agents in clinical trials, with tests currently available in clinical practice64,65,66. PD-L1 expression on TCs has been associated with immunosuppression, inhibiting the antitumor activity of T cells, as PD-L1 binding to PD-1 on activated T cells inhibits T-cell signaling and blocks antitumor immune responses27. PD-L1 expression alone may not be representative of the entire tumor microenvironment, as other concurrent immunosuppressive mechanisms, involving Treg, MDSC, TAM, and IDO, can be present64,65. Higher ORRs have been observed in patients positive than negative for PD-L1, although up to 17% of PD-L1 negative patients responded43,65,66,67,68. Moreover, about 50% of patients with high PD-L1 expression did not respond48,65. High PD-L1 expression has also been associated with longer PFS and OS, although the results varied among clinical trials64,65,66,67,68. Taken together, these findings show that PD-L1 IHC is insufficient as a predictive biomarker and has limitations. For example, multiple antibodies have been utilized in IHC assays and the interpretation of their results has not been standardized. Other limitations include tumor heterogeneity among different sections of the same sample or at different tumor sites, and dynamic changes in PD-L1 expression over time68. Several studies have compared different antibodies and IHC systems for PD-L1 tests. The Blueprint PD-L1 IHC assay comparison project revealed high concordance among the 28-8, 22C3, and SP263 assays, whereas fewer TCs were stained with the SP142 assay69. Another study found that the 28-8 and E1L3N assays were comparable, whereas fewer cells were stained using the 22C3 assay and the SP142 assay detected significantly lower PD-L1 expression in TCs70. High similarity among the different IHC assays suggests their potential interchangeability in clinical practice71. The U.S. Food and Drug Administration (FDA) has approved PD-L1 IHC 22C3 pharmDx as a companion diagnostic assay for pembrolizumab treatment in patients with NSCLC72.
2) Alternative biomarkers
Clinical trials have shown that melanomas and NSCLCs respond better to immune checkpoint inhibitors than other tumor types68. One study showed that melanomas and NSCLCs have the highest somatic mutation burden among tumor types73, suggesting that tumor mutation burden may be a predictive biomarker for treatment with immune checkpoint inhibitors. In patients with NSCLC who were treated with pembrolizumab, a higher nonsynonymous mutation burden was associated with higher ORR, longer PFS, and more durable clinical benefits74.
Many ongoing studies are attempting to identify new predictive biomarkers, including TILs, immune gene signatures, and multiplex IHC assays67. Understanding the tumor microenvironment and characterizing TILs and concurrent immunosuppressive mechanisms are therefore important.
Conclusion
Better understanding of the immune system in the tumor microenvironment has resulted in the development of new immunotherapy agents such as immune checkpoint inhibitors. Clinician familiarity with tumor immunology can help in understanding the process of immunotherapy and developing optimal treatment strategies for patients.
Immune checkpoint inhibitors, especially anti-PD-1 and anti-PD-L1, have become the standard of care in patients with advanced NSCLC. Nivolumab, pembrolizumab, and atezolizumab are safer and more effective than docetaxel. Pembrolizumab should be considered a first-line treatment in NSCLC with PD-L1 expression ≥50%, but without EGFR mutation and ALK rearrangement. Durvalumab may be an effective adjuvant treatment in stage III NSCLC after chemoradiotherapy.
Despite durable long-term survival, immune checkpoint inhibitors alone have a relatively low response rate. Therefore, combining immune checkpoint inhibitor with other immunotherapy or conventional therapy agents may improve response rates and provide clinical benefits to more patients. Predictive biomarkers are also essential in selecting patients who would benefit from treatment, reducing the unnecessary cost burden to patients who would not benefit from these agents. Although IHC assays of PD-L1 expression have drawbacks, they are the only currently available tests in clinical practice. Ultimately, biomarker-driven combination therapy will become a standard strategy in future immunotherapy.


 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Print
Print Download Citation
Download Citation