Introduction
The expansion of the elderly population has profound implications for society. In medicine, chronic diseases affect older adults disproportionately, rendering aging itself a root of disability, reduced quality of life and increased health care costs. As aging itself has emerged as the greatest risk factor for almost every major causes of morbidity and mortality in the modern world, efforts have been spurred to understand the molecular mechanisms which underlie the biology of aging and gain insights into potential interventions to delay aging and promote healthy longevity1.
Aging is thought to be caused by imperfections inherent in the living system which lead to progressive decline in fitness due to cumulative deleterious alterations of biological functions in the living system2,3. Similar to other organ systems, biological aging of the pulmonary system (lung aging) is associated with structural changes leading to a progressive decline in function4,5. These characteristics include (1) increased diameter of alveolar ducts and alveoli with loss of alveolar surface area, leading to diminished gas exchange; (2) decreased capillary density contributing to lower diffusion capacity; and (3) changes in inflammatory processes and alteration in immune cell functions4,5,6. The genetic, molecular, and cellular mechanisms involved in lung aging, however, are still poorly understood7,8.
Recently, an increasing body of evidence suggests that many of the important biologic features of aging biology are also found in lung aging, and that these changes contribute to the high incidence of lung diseases in the elderly9,10. In line with this, a joint workshop sponsored by the National Heart, Lung, and Blood Institute (NHLBI) and National Institute of Aging (NIA) of the United States asserted that enhanced understanding of how the aging process contributes to the development and/or the progression of lung diseases is urgently required8. In addition, it advocated for further research wherein biological hallmarks of aging in the lung are integrated with the pathobiology of lung diseases with divergent pathologies, for which advanced age is the most important risk factor8.
Numerous articles are available in which various aspects of aging biology are covered comprehensively. Avoiding an exhaustive summary, recent excellent reviews are referenced in the text. Instead, this review is constructed to serve the purpose of integrating recent molecular understanding of aging biology with pulmonary medicine. For this, first, current conceptual hallmarks of aging biology are briefly summarized. Then, recent scientific advancements obtained from studies of aging-associated lung diseases are interpreted through the prism of aging biology.
Hallmarks for Molecular Understanding of Aging Biology
Key features of aging have been conceptualized as ‘hallmarks of aging’ in order to capture the essence of molecular understanding of aging biology11. These conceptual hallmarks are genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication11.
Genomic instability refers to an increased tendency of alterations in the genome during the life cycle of cells. Since the first publication of Somatic Mutation Theory of Aging12, the DNA damage theory of aging, which argues that genomic instability plays a causal role in aging, has continuously propelled molecular understanding of aging biology13,14.
The telomere protects the genomic DNA through various mechanisms; thus, telomere attrition can lead to potentially maladaptive cellular changes, block cell division, and interfere with tissue replenishment. The roles of telomere biology, especially in diseases of human aging and in some aging-related processes, are well established15.
Epigenetic changes, resulting in alterations of gene expression and disturbances in broad genome architecture and the epigenomic landscape, are known to be associated with aging16. General loss of histones and the consequent transcriptional deregulation, imbalance of activating and repressive histone modifications, site-specific losses and gains of heterochromatin, alteration in DNA methylation levels and patterns are considered as evidence for epigenetic changes in aging17.
Eukaryotic protein homeostasis, or proteostasis, is maintained by a diverse and complex network of integrated functions, and overwhelming evidence supports the maintenance of cellular proteostasis as one of the key processes in ensuring longevity18. Currently, many age-related disorders are investigated in the context of proteostasis failure, given that the physiological deterioration of the proteostasis networks with age is an important factor for the development and/or progression of these aging-related disorders18.
Calorie restriction (CR), a nutritional intervention of reduced energy intake but with adequate nutrition, has been shown to extend the healthspan and lifespan in rodent and primate models19. In addition, a recent human study supports this notion20. Nutrient sensing pathways become deregulated and lose effectiveness with age; accumulating evidence reveals that various nutrient sensing pathways are fundamental to the aging process21,22.
A decline in mitochondrial quality and activity has been associated with normal aging and correlated with the development of a wide range of age-related diseases23. Contributing to other hallmarks of the aging process such as cellular senescence, inflammaging, and the age-dependent decline in stem cell activity, mitochondrial dysfunction is continuously being highlighted as a foundation of aging biology23,24,25.
Cellular senescence, a permanent state of cell cycle arrest that occurs in proliferating cells subjected to different stresses, is a state implicated in various physiological processes and a wide spectrum of age-related diseases26,27,28. In this regard, a pharmacologic approach called senotherapy, which refers to the selective killing of senescent cells by senolytic agents, has recently gained momentum to improve healthy aging and age-related diseases27,29.
The decline in the regenerative potential of tissues is one of the most obvious characteristics of aging, and stem cell exhaustion is widely accepted as a contributor to the decline in health during aging30. In accordance with this concept, aging phenotypes have been described for stem cells of multiple tissues, including those of the hematopoietic system, intestine, muscle, brain, skin, and germline. Various research efforts are currently being undertaken to test interventions that delay stem cell aging and improve both health and lifespan31,32.
Beyond cell-autonomous alterations, aging also involves changes at the level of intercellular communication which could be endocrine, neuroendocrine, or neuronal communication networks of an organism11. Obviously, an organism's health is orchestrated by a multitude of molecular and biochemical networks responsible for ensuring homeostasis within cells and tissues. Upon aging, a progressive failure in the maintenance of this homeostatic balance occurs in response to a variety of endogenous and environmental stresses, allowing the accumulation of damage, the physiological decline of individual tissues, and susceptibility to diseases33.
Of note, inflammaging, which refers to a low-grade proinflammatory phenotype that accompanies aging in mammals, is often remarked as an additional conceptual hallmark of aging34. Major age-related diseases share a common inflammatory pathogenesis, which is evidently seen in dementia, depression, atherosclerosis, cancers, diabetes and even mortality35,36. As such, “inflammaging” is often identified and investigated as an independent conceptual entity in aging biology.
Overall, these 10 conceptual hallmarks of aging are widely accepted to represent common denominators of aging, and provide a systemic perspective of how we can understand the molecular and cellular basis of aging biology.
Intersection of Aging Biology and the Pathobiology of Chronic Lung Diseases
Among the expert panels of aging biology researchers, there was a consensus in relation to which strategies could be promoted for the development of safe interventions to slow aging and increase healthy lifespan in humans37. These approaches comprise of (1) dietary interventions mimicking chronic dietary restriction (periodic fasting mimicking diets, protein restriction, etc.); (2) drugs that inhibit the growth hormone/insulin-like growth factor 1 (IGF-I) axis; (3) drugs that inhibit the mechanistic target of rapamycin (mTOR)-S6K pathway; (4) drugs that activate AMP-dependent kinase (AMPK) or specific sirtuins; (5) chronic metformin use; or (6) pharmacological inhibition of inflammation37.
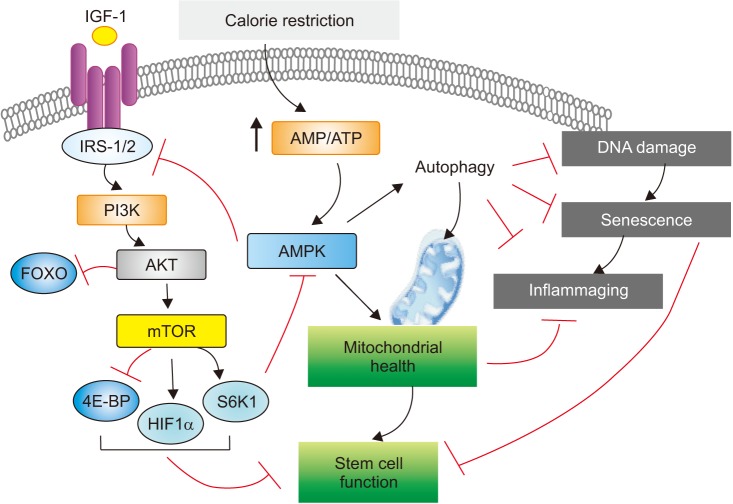
Of note, the molecular pathways targeted by these approaches are intricately connected with each other to regulate the hallmarks of aging biology. For example, CR is known to produce a pleiotropic effect and improves multiple metabolic pathways, generating benefits to the whole organism38,39. As illustrated in Figure 1, among the effects of CR, modulation of mitochondrial activity and a decrease in oxidative damage are well known. In addition, an intricate network of signaling pathways including IGF-1, mTOR, and AMPK molecules have been identified to mediate CR-induced beneficial effects on aging38,39. The anti-inflammatory effect of CR is also an interesting emerging factor to be taken into consideration38.
Given the significance of these molecules/signaling pathways in human aging, it is not surprising that the salient points related to each of the above strategies are continuously highlighted in the studies of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF), two exemplar diseases associated with aging. In this section, these recent findings are integrated into the context of aging biology, focusing on the strategies noted above.
1. Calorie restriction
To date, the most reliable, best-researched way to extend life span is through the practice of CR, which involves reducing calorie intake while simultaneously maintaining good nutritional status. Indeed, epidemiological and experimental data indicate that diet plays a central role in the pathogenesis of many age-associated chronic diseases, as well as in the biology of aging itself (see Figure 1). Human studies indicate that long-term CR with adequate intake of nutrients results in several metabolic adaptations that reduce the risk of developing type 2 diabetes, hypertension, cardiovascular disease, and cancer22.
Although the effects of diet on lung aging per se has thus far been rarely studied, several studies suggest a significant role of dietary modulation on lung aging. When a study examined the effects of aging on lung epithelial and stem cells and the effect of CR on young and old lungs, CR was identified to induce several potentially beneficial changes in lung epithelial cells, even when it is initiated at an older age, including reversal of some aging-induced changes40. Another study evaluated the effects of high-fat diet (HFD) in relation to lung aging41. As known, HFD decreases the lifespan of mice and is a risk factor for several human diseases. Indeed, this study revealed that HFD induces several histological, inflammatory, and functional changes in the lung, and exacerbates aging-induced lung inflammation and mitochondrial deterioration41. Of note, although a body of literature suggested the development of emphysema following severe CR and led to the notion of “nutritional emphysema” that might have relevance in COPD patients, neither the mechanics nor the histology showed any evidence of emphysema-like changes with severe CR in a murine study42.
Resveratrol (a substance found in red grapes and other plants), is one potential CR mimetics that may have beneficial effects against numerous diseases such as type 2 diabetes, cardiovascular disease, and cancer in tissue culture and animal models. Interestingly, a recent discovery suggested that the metabolic effects of resveratrol may be mediated by inhibiting cAMP phosphodiesterases (PDEs), particularly PDE443. Roflumilast, belonging to the class of PDE4 inhibitors, is currently used to treat COPD due to its ability to inhibit inflammatory cell responses44. Combined together, these findings may suggest that PDE4-related molecular pathways might be involved both in the biology of lung aging and COPD pathogenesis.
2. Growth hormone/IGF-1 axis
The growth hormone/IGF axis can be manipulated in animal models to promote longevity; IGF-related proteins including IGF-I and IGF-binding protein-3 (IGFBP-3) have also been implicated in risk of aging-associated diseases in humans45. Indeed, a recent study which evaluated lung function parameters in a large cohort of patients with acromegaly, revealed that these patients showed signs of small airway obstruction46. However, the idea of inhibiting the growth hormone/IGF-I axis for the management of COPD may not be straightforward. Many patients with COPD are malnourished, and this affects respiratory muscle function and prognosis for survival adversely. Previously, there were attempts to use recombinant human growth hormone treatment which has been proposed to improve nitrogen balance and to increase muscle strength in patients with COPD47, although significant beneficial effects were not observed48.
It is well established that multiple mesenchymal growth factors have been shown to be exaggerated in several fibrotic lung disorders including IPF, sarcoidosis, and bronchopulmonary dysplasia, as well as pulmonary manifestations of systemic diseases such as rheumatoid arthritis or progressive systemic sclerosis49. These growth factors include transforming growth factor (TGF)-β, IGF-I, platelet-derived growth factor, connective tissue growth factor, fibroblast growth factors, and keratinocyte growth factors. Another study demonstrated IGFBPs may play an important role in the development of fibrosis in IPF50.
3. mTOR signaling
A highly conserved protein kinase, mTOR (originally known as mammalian target of rapamycin), is continuously highlighted as a key modulator of aging in evolutionarily divergent organisms, ranging from yeast to rodents, and it is likely that this function has been conserved to some extent in humans51,52. A series of studies showed that rapamycin extended lifespan in yeast, nematodes, fruit flies and mice, firmly establishing mTOR signaling as a central, evolutionarily conserved regulator of longevity. Accordingly, much effort has been applied to defining the underlying mechanisms as to how mTOR signaling plays as a key modulator of aging-associated lung disorders.
Rtp801, a stress-related protein triggered by adverse environmental conditions and inhibits mTOR, has recently been identified to represent a major molecular sensor and mediator of cigarette smoke-induced lung injury53. In this study, Rtp801 mRNA and protein were overexpressed in human emphysematous lungs and in lungs of mice exposed to cigarette smoke. Mechanistic experiments found that Rtp801 was necessary and sufficient for nuclear factor-κB activation, alveolar inflammation, oxidative stress and apoptosis of alveolar septal cells, which have been identified to be key molecular mechanisms for COPD pathogenesis53. Demonstrating that mTOR activation was sufficient to induce lung cell senescence and mimic COPD lung alterations, with the rapid development of lung emphysema, pulmonary hypertension, and inflammation, the results from this study support a causal relationship between mTOR activation, lung cell senescence, and lung alterations in COPD54. In line with this, it was recently shown that lamin B1 protein levels are reduced in COPD lungs, contributing to the process of cigarette smoke (CS)-induced cellular senescence via dysregulation of mTOR and mitochondrial integrity55. Contrasting data exist, however. A study demonstrated that mTOR suppresses CS-induced inflammation through modulation of autophagy, apoptosis, and necroptosis, suggesting that activation of mTOR may represent a novel therapeutic strategy for COPD56. Another study suggested that particulate matter (PM) may inactivate mTOR signaling and promote the subsequent autophagy-mediated epithelial injury in PM-induced airway inflammation57. This apparent discrepancy may be due to the complex and heterogenic enzymatic pathway of mTOR signaling, implying that translation on the use of mTOR inhibitors to COPD therapy requires a more in-depth knowledge of mTOR signaling58.
Aging may affect adaptive responses to stress decreasing autophagy through activation of mTORC1 in lung fibroblasts, and this mTOR activation may contribute to the resistance to cell death in IPF lung fibroblasts59. In addition, a recent meta-analysis of genome-wide studies across three independent cohorts reported the importance of mTOR signaling in lung fibrosis60. Interestingly, by demonstrating that the alteration of canonical mTOR/phosphoinositide 3-kinase (PI3K)/AKT signaling is causally associated with increased Yes-associated protein (YAP) activity in respiratory epithelial cells in lungs of patients with IPF, a recent study expanded the functional significance of mTOR into Hippo/YAP signaling61. As known, Hippo/YAP pathway is a recently identified signaling cascade that plays an evolutionarily conserved role in the regulation of cell proliferation and differentiation during organogenesis and tissue repair62. Thus, YAP and mTOR/p-S6 signaling pathways may interact to induce cell proliferation and migration and inhibit epithelial cell differentiation that may contribute to the pathogenesis of IPF.
Of note, a recent study suggested that the mTORC1/4E-BP1 axis may represent a critical signaling node during fibrogenesis63. In this study, rapamycin-insensitive mTORC1 signaling via 4E-BP1 was shown to be a critical pathway for TGF-β1 stimulated collagen synthesis in human lung fibroblasts, whereas canonical PI3K/AKT signaling was not required63. This observation may have significant implication because, currently, pan-PI3K/mTOR inhibition is under clinical evaluation to test a therapeutic potential of targeting mTOR signaling in subjects with IPF64,65.
Taken together, alterations in mTOR signaling, which are associated with dysregulation of autophagy, inflammation, cell growth, and survival, may lead to the development of lung fibrosis66. Given complicated nature of mTOR signaling, it would be critical to continue basic research to elucidate this further and refine specific molecular targets involved in mTOR signaling for the development of novel IPF therapeutics.
4. AMPK signaling and sirtuins
Eukaryotes have evolved a very sophisticated system to sense low cellular ATP levels via the serine/threonine kinase AMPK complex. This energy switch controls cell growth and several other cellular processes, including lipid and glucose metabolism and autophagy, positioning the AMPK complex as a central mediator of the cellular response to energetic stress and mitochondrial insults leading to the coordination of multiple features of autophagy and mitochondrial biology67.
Sirtuins (SIRTs) are well-known mediators of aging68. Suppression of cellular senescence by SIRTs is mainly mediated through delaying age-related telomere attrition, sustaining genome integrity and promotion of DNA damage repair69. In addition, SIRTs modulate the organismal lifespan by interacting with several lifespan regulating signaling pathways including insulin/IGF-1 signaling pathway and AMPK69. For example, AMPK was known to control the expression of genes involved in energy metabolism by acting in coordination with another metabolic sensor, the NAD+-dependent type III deacetylase SIRT1, indicating the AMPK-induced SIRT1-mediated deacetylation may explain many of the convergent biological effects of AMPK and SIRT170. As such, recent data obtained from lung aging-associated disorders in relation to AMPK or SIRTs are discussed under the same sub-section, although the functional roles of these two molecules are not entirely dependent each other in the regulation of aging hallmarks.
SIRT1 has been identified to be reduced in lungs of patients with COPD71. In addition, SIRT1 activation attenuated stress-induced premature cellular senescence and protected against emphysema induced by CS and elastase in mice, implicating that activation of SIRT1 may be an attractive therapeutic strategy in COPD/emphysema72. Another member of the SIRT family, SIRT6, a histone deacetylase, antagonizes cellular senescence, through the attenuation of IGF-AKT signaling. In line with this, a study demonstrated that SIRT6 expression levels were decreased in lung homogenates from COPD patients, and SIRT6 expression levels correlated significantly with the percentage of forced expiratory volume in 1 sec/forced vital capacity, implicating that SIRT6 might be involved in COPD pathogenesis via its regulation of cellular senescence which can be attributed to IGF-AKT-mTOR signaling73.
A study suggested that accelerated epithelial senescence which can be antagonized by SIRT6 might play a role in IPF pathogenesis through perpetuating abnormal epithelialmesenchymal interactions74. When the mRNA and protein levels of all seven known sirtuins (SIRT1-7) were assessed in primary lung fibroblasts from patients with IPF and systemic sclerosis-associated interstitial lung disease in comparison with lung fibroblasts from healthy controls, these unbiased tests revealed a tendency for all SIRTs to be expressed at lower levels in fibroblasts from patients compared with controls, but the greatest decrease was observed with SIRT775.
Overall, SIRTs have been identified to regulate inflammation, aging (life span and health span), mitochondrial biogenesis, stress resistance, cellular senescence, endothelial functions, apoptosis/autophagy, and circadian rhythms through deacetylation of transcription factors and histones. Hence, various novel ways to activate sirtuins, either directly or indirectly, may have therapeutic potential in attenuating inflammation and premature senescence involved in chronic lung diseases76.
5. Metformin
Metformin has been shown to significantly increase lifespan and delay the onset of age-associated decline in several experimental models, and a growing body of evidence from clinical trials suggests that metformin can effectively reduce the risk of many age-related diseases and conditions, including cardiometabolic disorders, neurodegeneration, cancer, chronic inflammation, and frailty77. Regarding its pharmacologic action, it is important to note that metformin-induced activation of the energy-sensor AMPK is well documented, but may not account for all actions of the drug, requiring further elucidation of the underlying mechanisms of its action78.
COPD is often associated with type 2 diabetes mellitus (T2DM). Because metformin is a first-line treatment for most patients with T2DM, a study was aimed at investigating the effect of metformin on health care utilizations in patients with coexisting COPD and diabetes mellitus (DM). Interestingly, the use of metformin in patients with coexisting COPD and DM was associated with fewer COPD-specific emergency room visits and hospitalizations79. Another systematic review of pubic databases found also that metformin may benefit patients with COPD and T2DM by improving overall health status including symptoms, hospitalizations, and mortality80.
A therapeutic potential of metformin in IPF was implicated in a recent study81. This study demonstrated that, in humans with IPF and in an experimental mouse model of lung fibrosis, AMPK activity was lower in fibrotic regions associated with metabolically active and apoptosis-resistant myofibroblasts81. Pharmacological activation of AMPK in myofibroblasts from lungs of humans with IPF displayed lower fibrotic activity, along with enhanced mitochondrial biogenesis and normalization of sensitivity to apoptosis. In addition, in a bleomycin model of lung fibrosis in mice, metformin therapeutically accelerated the resolution of well-established fibrosis in an AMPK-dependent manner, further supporting a role for metformin to reverse established fibrosis by facilitating deactivation and apoptosis of myofibroblasts81. Similar evidence was provided by another report which demonstrated that metformin exerts potent antifibrotic effects in the lung by modulating metabolic pathways, inhibiting TGF-β1 action, suppressing collagen formation, and inducing lipogenic differentiation in lung fibroblasts derived from IPF patients82. When a post hoc analysis was undertaken to assess the effect of metformin on clinically relevant outcomes in patients with IPF, however, metformin showed no effect on clinically relevant outcomes in patients with IPF83.
6. Inflammaging, cellular senescence, and organismal aging
An age-associated increase in chronic, low-grade sterile inflammation termed “inflammaging” is a characteristic feature of mammalian aging that shows a strong association with occurrence of various age-associated diseases. Although the mechanism(s) responsible for inflammaging and its causal role in aging and age-related diseases are still elusive, emerging evidence suggests that one of basic processes that may contribute to age-related dysfunction and chronic sterile inflammation is cellular senescence84. Indeed, it has been demonstrated that chronic inflammation may derive, at least in part, from senescent cells: senescent cells secrete proinflammatory cytokines, chemokines, and proteases, termed the senescence-associated secretory phenotype84. In addition, age-associated accumulation of damage-associated molecular patterns has been proposed as a potential driver of inflammaging25,85.
Although it is not clearly defined whether the pulmonary environment becomes inflammatory with increasing age in humans, an in vivo study using a murine model organism suggests this possibility86. The lungs of old mice have elevated levels of proinflammatory cytokines and a resident population of highly activated pulmonary macrophages that are refractory to further activation by interferon-γ86. Another study suggested that inflammaging per se, which is induced over aging, could increase susceptibility to cigarette smoke-induced COPD87. The data from this murine experimental study revealed that age-induced lung inflammation is further elevated after CS exposure in old mice. An age-induced change in immune cells might play as a susceptibility factor to CS exposure, accelerating the pathophysiological hallmarks of COPD87.
Conclusion
Aging remains a grand mystery of biology. Although recent investigations have made key insights that provide molecular mechanisms underlying aging biology, our understanding remains elusive about how these age-related molecular and cellular alterations lead to the physiologic decline and loss of resilience at the level of an organ or an individual. This may be particularly true for the biology of lung aging. The body of scientific knowledge related to lung aging and its contribution to the pathobiology of chronic lung diseases is lagging far behind the recent advances in other areas of aging biology. As remarked at the recent NHLBI/NIA workshop, how scientific advances in molecular and cellular insights of aging biology can be integrated and constructed into a conceptual framework that describes mechanistic explanation of lung aging and its related chronic lung disorders represents a major challenge in pulmonary medicine8. The strong clinical association between advanced age and the steadily increasing morbidity and mortality attributable to chronic lung diseases signifies a fundamental link between the biology of aging and these lung diseases. Certainly, a much larger community of researchers or physician scientists beyond just “aging researchers” should be invited to ponder upon this mystery.
 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Print
Print Download Citation
Download Citation