Introduction
Guidelines are established to help medical staff select the best approach for individual patients: the medical staff considers whether the impact of clinical outcomes and the severity of the effects outweigh the risks of specific diagnoses or treatment methods. Several international and domestic academic societies have provided many guidelines for treating various diseases. However, the diagnosis and the treatment of pulmonary hypertension (PH) has mainly been performed by following European guidelines in Korea because the country lacks domestic guidelines. However, European treatment guidelines do not reflect the actual status of Korean patients. Therefore, new strategies of diagnosis and treatment should be fully developed for PH patients in Korea. Thus, we developed this guideline to help diagnose and treat PH appropriately in Korea, which is a country where the basis for the diagnosis and treatment of PH remains insufficient. The basic purpose of treatment guidelines is to assist medical staff in decision-making process through appropriate communication between physicians and patients in clinical practice. Our guideline is included in the first edition of guidelines for the diagnosis and treatment of PH in Korea. This edition is based on the ‘2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension’ with the acceptance and adaptation of recent publications of PH. Moreover, we tried to include in this edition as many articles as possible on Korean data [1].
The joint task force for the diagnosis and treatment of PH included the authors wrote and reviewed panels without conflict of interest. The panel of writers, which included five members of the Korean Society of Cardiology (KSC) and the Korean Academy of Tuberculosis and Respiratory Diseases (KATRD), created a draft that included an ‘up-to-date’ published evidence for the diagnosis, treatment, prevention, and rehabilitation of PH patients, including those living in Korea. The review panel comprised of 15 members, and they represented the opinions of 11 associated academic societies, including the Korean College of Rheumatology (KCR), the Korean Heart Rhythm Society (KHRS), the Korean Pediatric Heart Society (KPHS), the Korean Society of Heart Failure (KSHF), the Korean Society of Cardiovascular Imaging (KOSCI), the Korean Society of Echocardiography (KSE), the Korean Society of Interventional Cardiology (KSIC), the Korean Society for Transplantation (KST), the Korean Society for Laboratory Medicine (KSLM), the Korean Pulmonary Hypertension Society in Korean Society of Hypertension (KPHS/KSH), and the Korean Association of Clinical Cardiology (KACC). To avoid conflicts of interests, the task force that prepared guidelines for PH did not seek any financial support from related industries.
For the accommodation and adaptation of this guideline, we searched a total of 31,077 articles that were published across a 10-year period. These articles were obtained from the following three databases: MEDLINE, PubMed, and EMBASE. After evaluating these articles with K-AGREE II (Appraisal of Guideline for Research and Evaluation II), including six domains and 23 items, we finally selected the ‘2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension’ as a reference standard. Then, we partially adapted the ‘Guidelines for the Treatment of Pulmonary Hypertension (Japanese Circulation Society 2017/Japanese Pulmonary Circulation and Pulmonary Hypertension Society 2017)’ [1,2]. Thereafter, we reviewed consensus manuscripts and included them in this process. As shown in Tables 1 and 2, the level of evidence and the strength of recommendations for each item were graded and presented according to a predefined scale.
This guideline represents the official information on PH, which was presented by selected members of the KSC, KATRD, KPHS, and KPHS/KSH, and the guideline will be updated periodically. The full version of the PH guideline can be found at the official websites of the following academic societies: the KSC (http://www.circulation.or.kr), the KATRD (http://www.lungkorea.org), and the Korean Medical Guideline Information Center (https://www.guideline.or.kr/evaluation/index.php?sub=1). In this guideline, the recommendations mentioned were prepared to help medical experts make decisions in their daily work. The existing evidence of individual issues was evaluated and summarized in this guideline, but the final treatment decision for each patient had to be made by an attending physician. After carefully discussing with the patients and their caregivers. In addition, it is the responsibility of the individual medical practitioner to check related regulations such as various permits and insurance benefits that change concerning medications and devices when prescribing.
In the future, follow-up investigations should be conducted to verify whether the actual daily practice is performed in accordance with the recommendations of these PH guidelines. Apart from this process, it is necessary to create a series of virtuous cycles, including clinical research, guideline preparation, the dissemination, and the clinical application of these guidelines.
Definition and Classification of PH
1. To confirm the diagnosis of PH and to support treatment decisions, medical staff should perform right heart catheterization (RHC) only after ensuring that the patient is in resting position (The strength of recommendation I, The level of evidence C).
2. It is recommended that medical staff performs RHC at expert centers (The strength of recommendation I, The level of evidence B).
3. Vasoreactivity testing is recommended in patients with any of the following conditions: idiopathic pulmonary arterial hypertension (PAH) (IPAH), hereditary PAH (HPAH), and PAH. These conditions may be induced by drugs or toxins to help us detect patients who experienced long-term benefits with high doses of a calcium channel blocker (CCB, The strength of recommendation I, The level of evidence C).
1. Definition of PH
In the resting position, the normal value of mean pulmonary arterial pressure (mPAP) is 14±3 mm Hg; the upper limit of normal value is about 20 mm Hg, which is twice the standard deviation while assuming a normal distribution of pulmonary arterial pressure (PAP) [3]. Thus, PH is defined as an increase in mPAP to ≥25 mm Hg in the resting position. Moreover, mPAP should be determined by performing RHC [1,4].
Recently, the 6th World Symposia on Pulmonary Hypertension (WSPH) took place in Nice, France. The task forces presented their consensus opinion that PH should be defined as mPAP >20 mm Hg rather than mPAP ≥25 mm Hg because two standard deviations were above the normal mPAP value. Thus, PH is the threshold for abnormal PAP [5]. After holding an active debate over why the criteria for PH was changed to mPAP >20 mm Hg in this guideline, we retained the threshold of PH as mPAP ≥25 mm Hg. This is because additional research studies should be conduct to support these new criteria and the consensus of medical experts.
PH can be defined hemodynamically by using various combinations of following parameters: PAP, pulmonary artery wedge pressure, cardiac output (CO), diastolic pressure gradient, and pulmonary vascular resistance (PVR). Table 3 describes the different hemodynamic classifications of PH.
RHC should be performed to define PH and to assess the severity of PH and to check the acute vasoreactivity in expert centers. When RHC is performed in expert centers, both the complication rate and the mortality rate are very low at 1.1% and 0.06%, respectively [6]. Vasoreactivity testing is performed in patients with any of the following conditions: IPAH, HPAH, and PAH, which could be induced by drugs or toxins to detect patients who can have long-term benefits from high-dose of a CCB [7,8].
2. Classifications of PH
There are five clinical classifications of PH patients, with similar pathophysiological mechanisms, clinical presentation, hemodynamic profiles, and treatment strategies (Table 4) [1,9]. In PAH patients with an acute vasodilator response, the survival rate was dramatically improved when they treated with long-term CCB [7,8]. Therefore, it is necessary to identify these patients where pulmonary vasoconstriction plays an important role in PAH pathophysiology is vital. Thus, these PAH patients with long-term responders to CCBs were incorporated into the group 1.5. The causes of pulmonary veno-occlusive disease (PVOD) and pulmonary capillary hemangiomatosis (PCH) are similar, and these conditions are closely associated with PAH. Therefore, patients with PVOD or PCH were included in the group 1.6. Persistent pulmonary hypertension of the newborn which exhibits different patterns from conventional PAH, was classified into a separate disease category, which was included within the group 1.7.
Both congenital and acquired heart diseases causing post-capillary PH were included in the group 2. Patients with PAH associated with congenital heart disease are classified into four groups according to their clinical profiles: (1) Eisenmenger’s syndrome, (2) PAH associated with prevalent systemic and pulmonary shunts, (3) PAH with small/coincidental defects, and (4) PAH after defect correction [1,10].
Group 3 PH is due to lung diseases and/or hypoxemia. In this group, the common causes of group 3 PH are interstitial lung disease and chronic obstructive pulmonary disease (COPD). Severe PH is usually uncommon in patients with severe interstitial lung disease or COPD. However, severe PH can be associated with the combined emphysema/fibrosis syndrome.
Group 4 PH was changed to include chronic thromboembolic pulmonary hypertension (CTEPH), and other pulmonary artery obstructions, including pulmonary sarcoma or angiosarcoma, other intravascular tumors, arteritis without connective tissue disease, congenital pulmonary artery stenosis, and parasites [10].
Chronic hemolytic anemia is associated with an increased risk of PH. Pre-capillary PH associated with chronic hemolytic anemia is different from other types of PAH in terms of pathological characteristics (plexiform lesions), hemodynamic characteristics (low PVR and increased CO), and response to specific treatments for PAH (no response). This group was considered a different clinical condition, and the classification was changed from group 1 to group 5.
Epidemiology and Genetics of PAH
1. Epidemiology of PAH
The data pertaining to the global incidence of PH is insufficient. The prevalence of PAH in Europe is in the range 15-60 patients per million adult population, and the incidence of PAH in Europe is 5-10 cases per million adult population per year [1,11].
The epidemiologic data of PH in Korea is also scant. A retrospective study was based on the data collected from the Health Insurance Review and Assessment Service (HIRA) claim database (from 2008 to 2016): there were 1,307 newly diagnosed PAH patients in total. The mean age of diagnosis was 44±13 years. Among them, 69.3% of the patients were female, and 30.7% of the patients were male [12]. The incidence of PAH in Korea was 4.8 patients per million adult population per year. In total, 625 PAH patients were enrolled in the Korean Registry of Pulmonary Arterial Hypertension (KORPAH) study, which is the first prospective registry that included participants from four domestic societies (KSC, KCR, KPHS, and KATRD) [13]. The mean age of participants was 48±16 years. Among them, 503 (80.5%) of the patients were females. The incidence of PAH was 1.9 per million adult population per year [13]. This data showed that a low proportion of PAH patients who are diagnosed and treated remains low, indicating that a more-active patient screening programs should be more actively implemented in Korea. However, clinicians should interpret these data with caution because the frequency of RHC performance was very low (39.8%), and the degree of participation varied according to each academic society. Globally, systemic sclerosis is the most common cause of PAH. In Korea, it seems that many PAH patients have an associated complication of systemic lupus erythematosus.
The clinical pattern and the incidence of PAH changed according to the clinical situation of patients [14]. In the U.S. National Institutes of Health registry, which was first implemented in 1981, the mean age of patients with IPAH was 36 years. After including more PAH patients, the average age of patients is currently 50-65 years in this registry. The overall prognosis seems to have improved over a period of time.
2. Genetics of PAH
Mutations of bone morphogenic receptor protein 2 (BMPR2) are the most well-known genetic anomalies, and heterogynous BMPR2 mutations account for about 75% of familial PAH patients and approximately 25% of sporadic PAH patients [15]. The BMPR2 gene encodes a type 2 receptor of bone morphogenetic proteins, which control vascular cell proliferation. In the PILGRIM study, which examined the genetic mutation and the clinical features of BMPR2 were examined in 73 patients of Korea. The results indicated that 22% of patients with IPAH had mutations in BMPR2 gene [16]. The patients with BMPR2 mutations were relatively younger (27 years vs. 47 years, p=0.02) and had a higher mPAP than non-carriers (64 mm Hg vs. 51 mm Hg, p<0.05).
The heritable conditions of PVOD/PCH were caused by bi-allelic mutations of eukaryotic translation initiation factor 2 alpha kinase 4 (EIF2AK4), which encodes a serine-threonine kinase in all the eukaryotes [17].
Diagnosis of PH
1. Transthoracic echocardiography is a first-line noninvasive diagnostic test in clinically suspected PH cases (The strength of recommendation I, The level of evidence C).
2. Ventilation/perfusion lung scan should be performed in cases with unexplained PH to exclude CTEPH (The strength of recommendation I, The level of evidence C).
3. Computerized tomography (CT) pulmonary angiography is recommended to evaluate patients with CTEPH (The strength of recommendation I, The level of evidence C).
4. Biochemistry, hematology, immunology, human immunodeficiency virus (HIV) tests, and thyroid function tests are recommended to evaluate the specific associated condition of all the patients with PAH (The strength of recommendation I, The level of evidence C).
1. Diagnostic tests of PH
The diagnosis of PH was conducted in cases with a clinical suspicion of the disease; the symptoms or findings of physical examination caused clinical suspicion of PH. Although RHC is performed to determine hemodynamic criteria, it is also mandatory in the diagnosis of PH. Nevertheless, several diagnostic tests is necessary. Apart from determining the cause of PH, it is also essential to evaluate the severity of the disease in a PH patient. The diagnosis has to be performed systematically and consistently. To perform a comprehensive evaluation of the patient, physicians determined following parameters: functional classification, exercise capacity, echocardiographic findings, blood tests, and hemodynamic status [18].
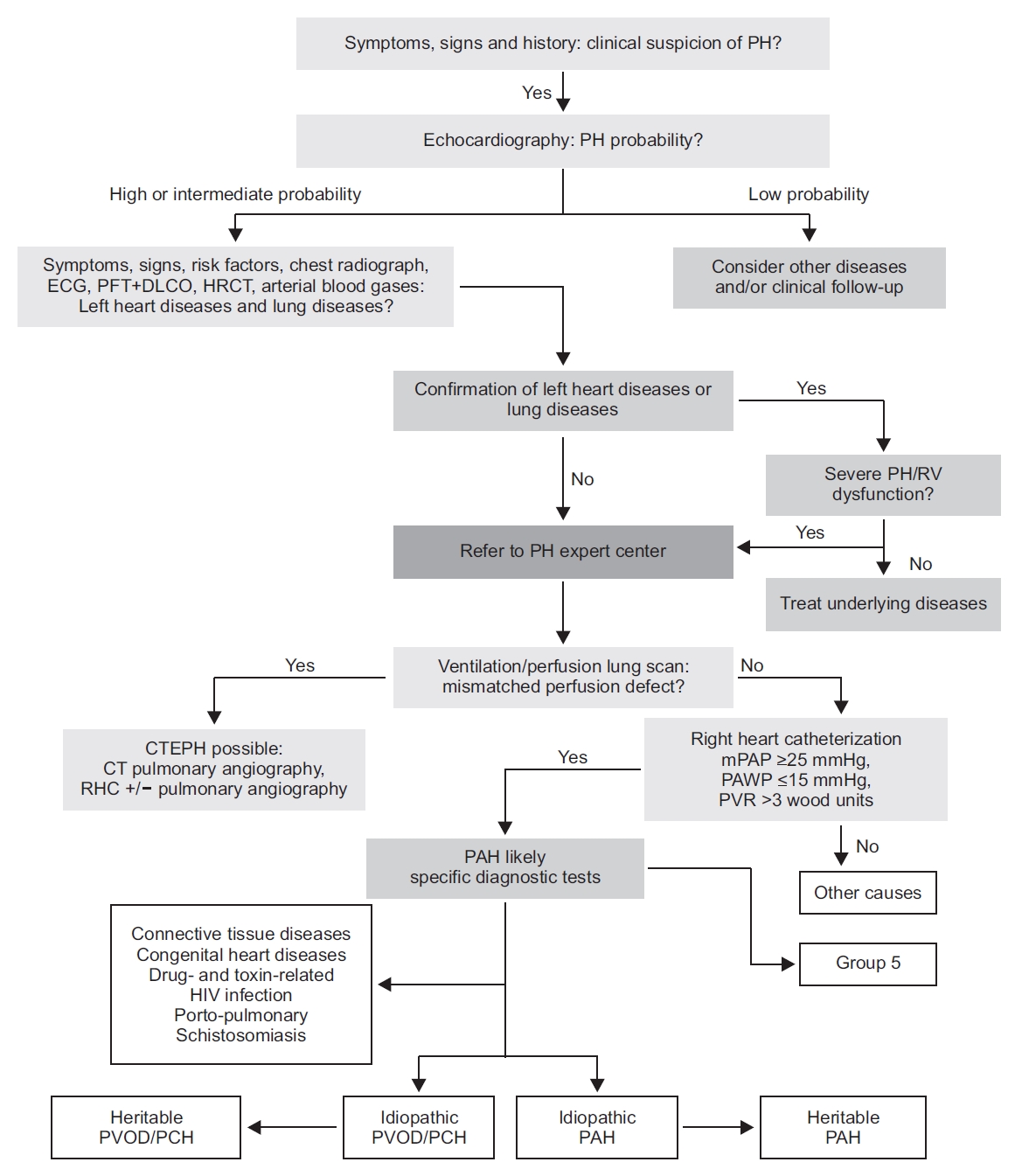
Transthoracic echocardiography is a first-line, non-invasive diagnostic test, which is performed on clinically suspected cases of PH. The probability of PH can be assessed by performing transthoracic echocardiography, which is a test that evaluates the effects of PH and estimates PAP with Doppler measurements [1,19]. When PH is probable, further evaluation tests such as RHC are recommended to confirm the diagnosis of PH.
In ventilation/perfusion lung scan, a normal or low probability result effectively excludes the possibility of CTEPH. Therefore, this test should be performed to evaluate CTEPH in patients with unexplained PH.
CT imaging can provide more important information on pulmonary vascular structure, and parenchymal and mediastinal abnormalities; these conditions may suggest the development of PH. To evaluate patients with CTEPH, we performed CT pulmonary angiography.
The biochemistry, hematology, immunology, HIV diagnosis tests, and thyroid function tests are recommended to evaluate the specifically associated condition of all patients with PAH.
2. Diagnostic algorithm
Figure 1 describes the diagnostic algorithm of PH. The suspected cases of PH were characterized by symptoms, signs, or clinical history. These patients had to undergo echocardiographic examination to determine the probability of PH. The more common clinical groups of PH (group 2 [PH due to left heart disease] and group 3 [PH due to lung disease and/or hypoxia]) had to be initially identified from patients with echocardiographic findings, which were compatible with PH. Then, group 4 (CTEPH) was distinguished by performing ventilation/perfusion lung scan on PH patients, which did not show any evidence of group 2 or group 3 PH. Furthermore, PH patients that could not be included in group 4 PH were classified by the presence of various causes of PAH (group 1) and rarer causes of PH (group 5).
Prognostic Evaluation and Risk Assessment of PAH
During the clinical evaluation of patients with PAH, it is necessary to provide valuable information pertaining to disease severity, improvement, deterioration, and stability. During follow-up visits, physicians note down basic history of patients, which includes changes in exercise capacity, episodes of chest pain, arrhythmia, hemoptysis or fainting, and changes in concomitant medications. In addition, physicians also evaluate whether the prescribed medications are compliant with the patient’s condition. By performing physical examination, physicians gather information on the presence or absence of distal or central cyanosis, edema, ascites, pleural effusion, and the dilatation of jugular vein. Moreover, they also measure the heart rate, rhythm, and blood pressure of PAH patients.
Although there are considerable differences among clinicians [20], the evaluation of dyspnea is performed according to the World Health Organization (WHO) functional class criterion as it is the most powerful indicator that predicts clinical prognosis during a follow-up visit and at the time of diagnosis.
The 6-minute walking test (6MWT), which evaluates the submaximal exercise capacity of a patient, is the most widely used exercise test at many PH centers. This test is easy to perform, inexpensive, and familiar to both the patients and the medical staff. Just like other PH assessments, physicians always interpret the results of 6MWT in clinical context. In general, the cardiopulmonary exercise test (CPET) is performed to evaluate maximal exercise capacity of PH patient. Thus, it provides important information on gas exchange, ventilatory efficiency, and cardiac function during the time of exercise, so it is used to evaluate the exercise capacity of a patient. It offers information on the end-tidal partial pressure of carbon dioxide (PCO2), ventilatory equivalents of carbon dioxide (VE/VCO2), oxygen/pulse (VO2/HR), and peak oxygen uptake (peak VO2). It was found that PAH patients had low PCO2, high VE/VCO2, low VO2/HR, and low peak VO2 [21].
Although various biochemical markers have been studied, physicians have not found a single specific indicator that reflects the degree of pulmonary vascular remodeling or the severity of PAH. Among these biomarkers, B-type natriuretic peptide (BNP) and N-terminal pro-BNP (NT-proBNP) concentrations correlate with myocardial dysfunction. Thus, they provide prognostic information both during the follow-up visits and at the time of diagnosis [22]. But, they are nonspecific biomarkers of PH and are associated with many cardiovascular diseases. Therefore, these levels of biomarkers should be interpreted in the clinical context.
Traditionally, risk assessment of PAH patients was performed in a multidimensional setting by performing imaging studies and by examining hemodynamic profiles. Thus, physicians consider the following parameters: symptoms, the WHO functional class, the clinical signs of right heart failure, the results of 6MWT or CPET, the expression of BNP/NT-proBNP, the presence of pericardial effusion, and the size of right atrial area. Since these variables are partially overlapping, it is difficult to measure all the variables at each follow-up visit. Therefore, a simplified evaluation system was presented at the sixth WSPH. Since it facilitated risk assessment, we adapted and included it in our guideline (Table 5) [23]. This is because it is unnecessary to evaluate all the parameters of PAH patients at every follow-up visit. However, clinicians should determine the functional class and check the patient’s exercise capacity by conducting 6MWT or CPET. It is also recommended to measure the biomarkers, BNP/NT-proBNP. Moreover, clinicians should also obtain information on the right ventricular function by performing an echocardiographic examination of PAH patients.
However, in the risk assessment proposed above, most of the variables are based on the published data and expert opinion. Therefore, clinicians should exercise caution while applying these variables to individual patients.
Treatment of PH
1. CCBs are recommended in patients with IPAH, HPAH and drug-induced PAH who have a positive vasoreactivity test (The strength of recommendation I, The level of evidence C).
2. Initial monotherapy or combination therapy of approved drugs should be provided to treatment-naïve patients with PAH and to patients with low- or intermediate-risk of PAH (The strength of recommendation I, The level of evidence A).
3. A sequential combination therapy of approved drugs is recommended in PAH patients who show an inadequate treatment response to initial monotherapy or initial double combination therapy (The strength of recommendation I, The level of evidence B).
4. The use of PAH-approved drugs is not recommended to PH patients with left heart disease or lung diseases (The strength of recommendation III, The level of evidence C).
5. Surgical pulmonary endarterectomy and deep hypothermic circulatory arrest is recommended for CTEPH patients with operability (The strength of recommendation I, The level of evidence B).
6. Riociguat is recommended in symptomatic CTEPH patients with persistent or recurrent lesions after surgical treatment or in inoperable CTEPH patients (The strength of recommendation I, The level of evidence B).
Initially, PAH patients should be managed with following general measures: physical activity, supervised rehabilitation, inhibition of pregnancy, birth control pills and post-menopausal hormonal therapy, prompt preprocedural management of elective surgery, prevention of infection, psychosocial support, adherence to treatments, genetic counseling, and recommended precautions during traveling. Supportive therapy for PAH patients includes oral anticoagulants, diuretics, oxygen, and digoxin.
Drug treatment for PAH patients should be provided on the basis of diagnosis, symptoms, and results of acute vasoreactivity test. Without contraindication to using CCBs, CCBs should be considered for PAH patients who show a favorable response to acute vasoreactivity test, which is performed along with RHC at the time of diagnosis. The choice of CCB is based on the heart rate at baseline: nifedipine (120-240 mg/day) and diltiazem (240-720 mg/day) are the CCBs that are frequently prescribed to PAH patients [7,8].
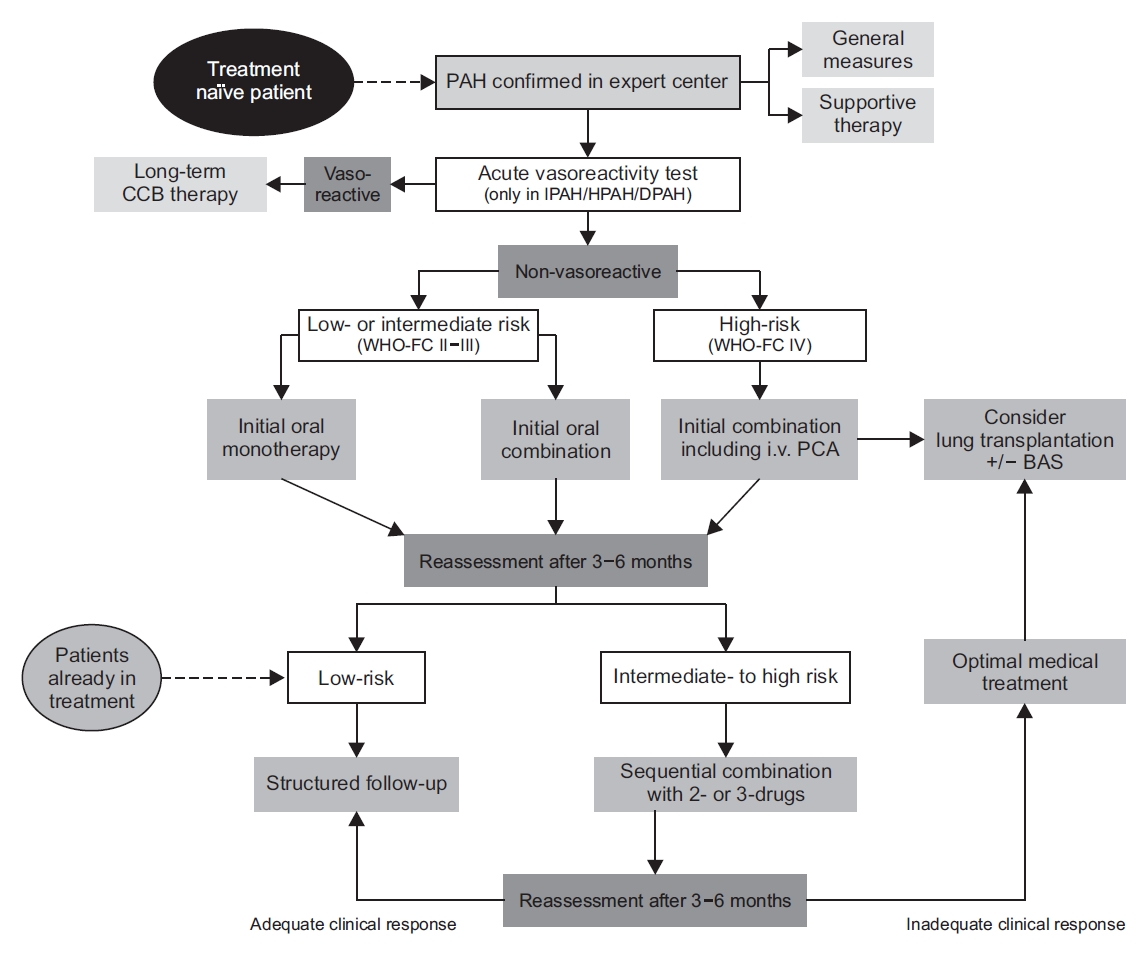
When PAH patients do not show vasoreactivity, clinicians can prescribe PAH-specific drugs according to the grade of recommendation and the level of evidence for treatment. However, it is not necessary to prescribe PAH-specific drugs to PAH patients who do not show any symptoms (WHO functional class I). A close follow-up is required to determine if and when symptoms occur in these patients and before deciding whether PAH-specific drugs should be prescribed [18]. For PAH patients in WHO functional class II or higher, the treatment is according to their individual indications. Figure 2 shows the treatment algorithm.
Clinicians prescribed the following PAH-specific drugs: endothelin receptor antagonists, phosphodiesterase type-5 inhibitors (PDE-5I), guanylate cyclase stimulators (cGC), prostanoids, and IP-receptor agonists. An initial monotherapy or combination therapy of PAH-specific drugs was recommended to newly diagnosed PAH patients and to patients with low- or intermediate-risk of PAH but without vasoreactivity. An initial combination therapy included intravenous prostacyclin analogs, which is prescribed to PAH patients of WHO functional class IV. An intravenous epoprostenol is the first line of treatment because it has reduced the initial mortality of high-risk PAH patients. However, this drug is not available in Korea (as of October 2020). Therefore, it is necessary to actively implement other types of initial upfront combination therapy. After providing initial monotherapy or initial double combination therapy, the treatment response is inadequate in the reassessment period of 3-6 months, then clinicians have to administer a sequential double or triple combination therapy of approved drugs to symptomatic PAH patients. The combination of PDE-5I and cGC drugs shows contraindications.
Pulmonary endarterectomy (PEA) in deep hypothermic circulatory arrest is recommended for CTEPH patients with operability assess by a multidisciplinary team. Riociguat can be prescribed to inoperable CTEPH patients or in patients with persistent or recurrent lesions after undergoing PEA. Moreover, balloon pulmonary artery angioplasty may be an intervention for inoperable CTEPH patients.
Even after providing initial monotherapy or combination therapy to PAH patients, the clinical response may be inadequate. In such a situation, PAH patients must be further examined to determine whether they are suitable candidates for lung transplantation.
Conclusion
Over the past decades, there have been major advances in the management of PH. However, the morbidity and mortality of PH is still significant. Moreover, the strategy used for the diagnosis and treatment of PH is mainly associated with European guidelines, even in Korea. This is because the country lacks localized guidelines for the diagnosis and treatment of PH. Since European treatment guidelines do not reflect the actual status of Korean patients, the diagnosis and treatment have not been effective on Korean patients with PH. Therefore, we developed a new guideline to help diagnose and treat PH cases appropriately in Korea, which is a country with insufficient data for the diagnosis and treatment of PH.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Print
Print Download Citation
Download Citation